
Official address Domenico Scarlalaan 6 ● 1083 HS Amsterdam ● The Netherlands
An agency of the European Union
Address for visits and deliveries Refer to www.ema.europa.eu/how-to-find-us
Send us a queson Go to www.ema.europa.eu/contact
Telephone +31 (0)88 781 6000
© European Medicines Agency, 2024. Reproducon is authorised provided the source is acknowledged.
28 June 2024
EMA/923413/2022 – v. 4.0
Clinical Trials Informaon System (CTIS) Programme
Clinical Trials Informaon System (CTIS) - Sponsor Handbook
A compilaon of key guidance, technical informaon, recommendaons, and references for
geng ready for the use of CTIS
Execuve summary
The aim of the EMA CTIS Sponsor Handbook (‘Handbook’) is to provide clinical trial (CT) sponsors
represenng pharmaceucal industry, SME (small and medium-sized enterprises), academia, research
organisaons and other clinical trial sponsor organisaons with the informaon they need to navigate
the Clinical Trials Informaon System (CTIS) - to create and submit clinical trial informaon to the
member states of the European Union as required by the Clinical Trial Regulaon [CTR: Regulaon (EU)
No 536/2014]. The Regulaon harmonises the assessment and supervision processes for clinical trials
throughout the EU/EEA, via CTIS. CTIS contains the centralised EU portal and database for clinical trials
foreseen by the Regulaon.
The Handbook addresses key quesons on CTIS and provides a compilaon and references to key
guidance, technical informaon, recommendaons, training materials, and supporve documentaon to
facilitate the submission and assessment of CTAs and addional informaon during the lifecycle of a trial.
It has been developed by the European Medicines Agency (EMA) in collaboraon with representaves of
industry stakeholders.
The Handbook will be revised as more informaon becomes available, or system funconalies are
updated. It is best used in conjuncon with the many references to which it points, for example, Volume
10 of the publicaon ‘The rules governing medicinal products in the European Union’ that contains
guidance documents applying to clinical trials (EudraLex - Volume 10 - Clinical trials guidelines).

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 2/62
Document evoluon
Version
Version descripon
Date
1.0
This first version of the CTIS Sponsor Handbook contains
priorised topics. Addional topics will be inserted/completed
in the document and updates will be provided in the next
versions.
28 July 2021
2.0
Updated handbook secons:
- Editorial changes across the document
- OMS registraon process (secon 3.2.1) updated
- User personas and organisaon models (secon 4.5) updated
with new links
- Product management in CTIS (secon 5) updated
- Transion from Direcve to Regulaon (secon 6) updated
- Data fields and documents specificaons (secons 7.1.3) new
- SUSARs reporng (secon 8.1) updated
- Training environment for user training and organisaon
preparedness (secon 10.4) new
30 November
2021
3.0
This document has been revised to its third version since CTIS
went live.
Updated handbook secons follow:
- Editorial changes and reference updates across the document
- Execuve summary updated
- Overview of Clinical Trial Applicaon (CTA) process in CTIS –
from submission to decision and reporng (secon 1.2) updated
- CTIS go-live date (secon 1.3) updated
- Organisaon and Sponsor Administrator registraon (secon
2.2) updated
- Key user management concepts in CTIS (secon 3.1) updated
29 November
2022

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 3/62
Version
Version descripon
Date
- Markeng Authorisaon Holder (MAH) group of users (secon
3.5) new
- How to get a clinical trial applicaon started in CTIS (secon 4)
new
- Transion from Direcve to Clinical Trial Regulaon (secon 5)
updated
- How to create a transional trial in CTIS (secon 5.3) new
- How to manage trials transioned to the CTR in CTIS (secon
5.4) new
- Data, documentaon, and processes (secon 7) updated
- Safety reporng obligaons (secon 9) updated
- Support (secon 10) updated
- Other references (secon 11) updated
3.01
- Minor editorial changes
- Update of melines due dates in CTIS in Secon 4.1
- Update of document upload size (secon 7.1.3.2)
- Clarificaons on dates for the transion period (secon 5.1,
5.2.2)
December 2022
3.02
- Recording sites locally in CTIS (secon 2.2.2) new
- CTIS Bitesize Talks links (mulple secons) new
- Mul-factor authorisaon in CTIS (secon 2.1.1) new
- Mulple Q&A reference links added (secon 10.6) updated
- Glossary (secon 12) updated
April 2023
3.03
- Module 7 video links (secon 2.2.3) updated
- Note added to Part II document 'Proof of insurance cover or
idenficaon' (secon 4.2, table 4.2.5) updated
- Addional reference materials for CTIS users link (secon 4.2)
updated
October 2023

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 4/62
Version
Version descripon
Date
- CTCG guidance on transional trials link (secon 5.1) updated
- Added link to CTCG Cover leer template for transional trials
(secon 5.1) new
- added link to the Guidance for the Transion of clinical trials
from the Clinical Trials Direcve to the Clinical Trials Regulaon
new
- Link Eudralex 10 page - Set of documents applicable to clinical
trials authorised under Regulaon EU No 536/2014 (secons
5.1 and 9.1) update
- Maximum number of documents upload in one batch (secon
7.1.3.2, table 7.1.3.2.1) new
- CT Highlights newsleer subscripon link (secon 10.2)
updated
- CTIS online training modules page (secon 10.4) updated
- CTIS Training Environment Support Service link (secon 10.5)
updated
- Added link to Q&A on protecon of CCI and personal data in
CTIS (secon 10.6) updated
- added link to Conclusion of VHP Procedure (secon 11) update
- ACT EU website link added (secon 11) new
- Reference to EMA Medical Terms Simplifier added (secon 12)
new
4.0
- Multi-factor Authentication (MFA) in CTIS (section 2.1.1)
updated
- Transition Period (section 5.1) updated
- Reference document to Guidance for the Transition of
clinical trials from the Clinical Trials Directive to the Clinical
Trials Regulation (section 5.1) updated
- Trials that should not be transitioned (section 5.2.1) updated
- Links throughout the document updated
June 2024

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 5/62
Table of Contents
Execuve summary ............................................................................................................................ 1
Table of Contents ............................................................................................................................... 5
1. What CTIS is and what it does ......................................................................................................... 7
1.1. A brief introducon to CTIS ..................................................................................................................................... 7
1.2. Overview of Clinical Trial Applicaon (CTA) process in CTIS – from submission to decision and reporng ............ 8
1.3. CTIS go-live date ...................................................................................................................................................... 9
2. Geng access to CTIS – registraons ............................................................................................. 10
2.1. User self-registraon ............................................................................................................................................. 10
2.1.1. Mul-factor authencaon in CTIS .................................................................................................................... 11
2.2. Organisaon and Sponsor Administrator registraon ........................................................................................... 11
2.2.1. Organisaon registraon in OMS ....................................................................................................................... 11
2.2.2. Organisaon registraon locally in CTIS for use in CTIS ...................................................................................... 13
2.2.3. Sponsor Administrator registraon in EMA Account Management portal for use in CTIS ................................. 13
3. Management of users and organisaons in CTIS ............................................................................ 15
3.1. Key user management concepts in CTIS ................................................................................................................ 15
3.2. User roles concept in CTIS ..................................................................................................................................... 15
3.3. Organisaon-centric approach - Sponsor Administrator ....................................................................................... 16
3.4. Trial-centric approach – Clinical Trial Administrator .............................................................................................. 17
3.5. Markeng Authorisaon Holder (MAH) group of users ........................................................................................ 18
3.6. CTIS user personas and organisaon models ........................................................................................................ 18
4. How to get a clinical trial applicaon started in CTIS ...................................................................... 19
4.1. Introducon ........................................................................................................................................................... 19
4.2. Draing of the clinical trial applicaon dossier ..................................................................................................... 19
4.3. Clinical Trial Applicaon under evaluaon ............................................................................................................ 24
4.4. Clinical Trial Applicaon aer decision .................................................................................................................. 24
5. Transion from Direcve to Clinical Trial Regulaon ...................................................................... 25
5.1. Transion period .................................................................................................................................................... 25
5.2. Points to consider on transional arrangements .................................................................................................. 27
5.2.1. What trials should not be transioned ............................................................................................................... 27
5.2.2. Can the trial be transioned? ............................................................................................................................. 27
5.2.3. What are the assessment melines of transional trials ................................................................................... 29
5.3. How to create a transional trial in CTIS ............................................................................................................... 29
5.4. How to manage trials transioned to the CTR in CTIS ........................................................................................... 29
6. Product management in CTIS ........................................................................................................ 30
6.1. Medicinal product registraon in XEVMPD ........................................................................................................... 30
6.2. Medicinal product in CTIS extracted from XEVMPD .............................................................................................. 32
6.3. Adding an authorised medicinal product in CTIS ................................................................................................... 32
6.4. Adding an unauthorised medicinal product in CTIS .............................................................................................. 34
6.5. Medicinal product details in CTIS .......................................................................................................................... 35

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 6/62
7. Data, documentaon and processes .............................................................................................. 36
7.1. Clinical Trial Applicaon (CTA) and Noficaon Forms .......................................................................................... 36
7.1.1. Clinical Trial Applicaon Form overview of the data fields to be completed and documents to be provided ... 36
7.1.2. Noficaon and Results: overview of the data fields to be completed and documents to be provided ........... 37
7.1.3. Data fields and document specificaons ............................................................................................................ 37
7.2. Trial categorisaon ................................................................................................................................................ 39
7.3. Download opons ................................................................................................................................................. 39
7.3.1. Clinical trial page ................................................................................................................................................ 39
7.3.2. Clinical trial applicaon page .............................................................................................................................. 40
7.3.3. Clinical Trial Search Download of results ............................................................................................................ 41
7.4. How to apply a change to a clinical trial dossier.................................................................................................... 42
7.4.1. During the draing of the inial clinical trial applicaon ................................................................................... 42
7.4.2. Once the inial clinical trial applicaon has been submied ............................................................................. 42
7.5. Handling of Requests for Informaon (RFIs) in CTIS .............................................................................................. 45
7.6. Subming the Clinical Study Report (CSR) ............................................................................................................ 49
8. Data transparency ........................................................................................................................ 49
9. Safety reporng obligaons .......................................................................................................... 51
9.1. Suspected Unexpected Serious Adverse Reacons (SUSARs)................................................................................ 51
9.2. Annual Safety Report (ASR) ................................................................................................................................... 52
10. Support ...................................................................................................................................... 53
10.1. Release notes and known issues ......................................................................................................................... 53
10.2. CTIS Highlights Newsleers ................................................................................................................................. 53
10.3. CTIS informaon events....................................................................................................................................... 54
10.4. CTIS training ......................................................................................................................................................... 54
10.5. CTIS training environment for user training and organisaon preparedness ...................................................... 55
10.6. Quesons and answers on CTR, CTIS and other EMA IT systems ........................................................................ 56
10.7. Support for SME and academia sponsors ............................................................................................................ 57
11. Other references ......................................................................................................................... 59
12. Acronyms and Glossaries ............................................................................................................ 60

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 7/62
1. What CTIS is and what it does
1.1. A brief introducon to CTIS
CTIS is the single-entry point for clinical trials informaon in the European Union (EU) and in the
European Economic Area (EEA).
This includes a single clinical trial applicaon dossier, covering clinical trial applicaons submied to
EU/EEA Member States, including submission to Naonal Competent Authories (NCAs) and Ethics
commiees (ECs) and registraon of the clinical trial in a public register; all in one integrated submission.
CTIS provides harmonised and simplified end-to-end electronic applicaon procedures over the lifecycle
of clinical trials across the EU/EEA.
CTIS is, however, not a clinical trial management system. It should therefore not be relied upon by
sponsors to store informaon on a clinical trial. CTIS provides a digital secured archive of documents,
decisions, and informaon on a clinical trial, but sponsors should ensure they ulise their own
informaon management system to store informaon needed for compliance purposes.
The exchange of informaon between sponsors and Member States is fully electronic in CTIS.
In CTIS, Member States collaborate and coordinate amongst themselves for the evaluaon and
supervision of clinical trials resulng in one single decision per Member State Concerned.
Documents can be uploaded but not created in CTIS.
CTIS offers searchable clinical trial informaon to the paent, the healthcare professional, and the
general public. Clinical trial results are available both as a technical summary and in lay language.
Informaon can be retrieved by searching for a parcular trial or across trials for treatment-related
details.
Paent safety in clinical trials is enhanced as CTIS provides an end-to-end electronic soluon for safety
reporng of trials.
CTIS facilitates a harmonised safety assessment in Europe, supported by agreed assessment report
templates.
The clinical trial module of EudraVigilance provides for the electronic reporng of Suspected Unexpected
Serious Adverse Reacons (SUSARs) by sponsors and re-roung to Member States.
CTIS delivers an electronic Annual Safety Reports (ASRs) repository.
Digitalisaon
& Improved
Efficiency
Increased
Transparency
Enhanced
Paent

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 8/62
CTIS is a unique intuive tool that facilitates the submission of clinical trial applicaons including those
for mul-naonal trials and therefore facilitang invesgaon of e.g. rare diseases. It thereby also
supports academic innovave work.
CTIS offers search and export of structured clinical trial data to allow efficient reporng for sciensts.
A clinical trial can be extended to more Member States e.g. to enhance recruitment rates.
1.2. Overview of Clinical Trial Applicaon (CTA) process in CTIS – from submission to decision and
reporng
CTIS is structured in two restricted and secured workspaces (Sponsor and Authority), only accessible to
registered users, and a website openly accessible to the general public.
The sponsor workspace provides clinical trial sponsors with the funconalies for submission of CTAs to
Member States, and management of informaon throughout the lifecycle of clinical trials.
The sponsor funconalies include:
• assignment and management of users;
• compilaon of clinical trial dossiers;
• receiving of alerts and noces for ongoing trials;
• compilaon of responses to requests for informaon;
• view deadlines, search, and access clinical trials;
• compilaon of noficaons related to the life cycle of the trial including submission of a summary of
clinical study results.
Registered users with the appropriate permissions are able to access the following tabs for their affiliated
trials:
The clinical trials tab provides search funconalies that facilitate users to find specific trials and view
informaon (see module 9 of the CTIS training programme).
The noces and alerts tab shows the messages triggered by acvies that occur during the life cycle of a
clinical trial (see module 4 of the CTIS training programme).
The tab for requests for informaon –RFI– tab provides access to such requests made by Member States
Concerned (MSC) for clinical trials, and enables users to view their status, due dates, and other relevant
informaon (see modules 4, 5 and 11 of the CTIS training programme).
The User administraon tab allows management of roles and permissions for all users that are registered
in the system (see modules 3 and 7 of the online CTIS training programme).
The CTIS Training Material Catalogue Module 2 provides a high-level overview of CTIS workspaces.
Support to
Innovaon &
Research

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 9/62
References
Locaon (area or document)
Clinical Trial Regulaon (EMA website)
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-trials/clinical-
trials-regulaon
CTIS Training Material Module 02 ‘High-
level overview of CTIS workspaces and
common system funconalies: User
Quick Guide’
hps://www.ema.europa.eu/documents/other/quick-
guide-overview-cs-workspaces-common-system-
funconalies-cs-training-programme-module-
02_en.pdf
CTIS Training Material Module 02 ‘High-
level overview of CTIS workspaces and
common system funconalies: CTIS
common funconalies, Part A’
hps://youtu.be/EgIRpL17WaU
CTIS Training Material Module 03 ‘User
access management’
hps://www.ema.europa.eu/documents/other/quick-
guide-user-access-management-cs-training-programme-
module-03_en.pdf
CTIS Training Material Module 04 ‘Support
with workload management - Noces &
Alerts’
hps://www.ema.europa.eu/en/learning-
module/workload-management-sponsor/story.html
CTIS Training Material Module 04 ‘Support
with workload management -RFIs’
hps://www.youtube.com/watch?v=6z-Q6-LK8Ws
CTIS Training Material Module 05 ‘Manage
a clinical trial through CTIS’
hps://www.ema.europa.eu/en/learning-
module/manage-ct/story.html
CTIS Training Material Module 09 ‘Search,
view and download informaon on clinical
trials and clinical trial applicaons: User
Quick Guide’
hps://www.ema.europa.eu/en/documents/other/quick-
guide-how-search-view-download-clinical-trial-clinical-
trial-applicaon-sponsors-cs_en.pdf
CTIS Training Material Module 11
'Respond to requests for informaon
received during the evaluaon of a clinical
trial applicaon'
hps://www.ema.europa.eu/documents/other/step-
step-guide-how-respond-requests-informaon-received-
during-evaluaon-clinical-trial_en.pdf
1.3. CTIS go-live date
CTIS was launched on 31 January 2022, and the Clinical Trial Regulaon has been applicable since then.
In April 2021, the EMA's Management Board confirmed that the system met the agreed requirements
following an independent audit of CTIS. On 31 July 2021, the European Commission confirmed 31 January
2022 as the date of entry into applicaon of the Clinical Trials Regulaon, and the go-live of CTIS, via
publishment of a noce in the Official Journal of the European Union.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 10/62
Since the launch of CTIS, there has been a 3-year transion period; this is covered in more detail in
Secon 5 ‘Transion from Direcve to Clinical Trial Regulaon’.
References
Locaon (area or document)
Informaon on clinical trials in the
context of Regulaon EU No 536/2014,
including the noce published in the OJ
(European Commission website)
hps://ec.europa.eu/health/human-use/clinical-
trials/regulaon_en
Informaon in the context of Regulaon
EU No 536/2014 (EMA website)
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-trials/clinical-
trial-regulaon
2. Geng access to CTIS – registraons
2.1. User self-registraon
In order to access the CTIS Sponsor workspace, a user needs to have an acve EMA Account.
If the user already uses other EMA applicaons (e.g. Eudralink, SPOR, IRIS, EudraVigilance, OMS), the
user already has an EMA Account and could access the CTIS Sponsor workspace using his/her exisng
EMA Account credenals.
If the user does not have an acve EMA Account, (s)he needs to create one, by self-registraon.
The self-registraon process is described on the EMA Account Management (IAM) homepage and in
Module 03 of the CTIS Training Material Catalogue.
References
Locaon (area or document)
EMA Account Management homepage
hps://register.ema.europa.eu/identyiq/home.html
Geng Started with CTIS: Sponsor Quick
guide
hps://www.ema.europa.eu/documents/other/geng-
started-cs-sponsor-quick-guide_en.pdf
CTIS Training Material Module 03 ‘User
Access Management: Quick Guide’
hps://www.ema.europa.eu/en/documents/other/quick-
guide-user-access-management-cs-training-programme-
module-03_en.pdf
CTIS Training Material Module 03 ‘User
Access Management: Frequently Asked
Quesons (FAQs)’
hps://www.ema.europa.eu/en/documents/other/faqs-
user-access-management-cs-training-programme-
module-03_en.pdf
CTIS Training Material Module 03 ‘User
Access Management: Videoclip’
hps://www.youtube.com/watch?v=VSLYv9l-LcE&ab

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 11/62
2.1.1. Mul-factor authencaon in CTIS
The mul-factor authencaon (MFA) strategy for user logins to CTIS, for both Sponsor and Member
State workspaces, was launched on 1 June 2023. This process reinforced the security of CTIS user
accounts.
For the MFA, it is recommended that each user is equipped with a mobile or an office phone that can be
used for second factor authencaon. Instrucons on seng up MFA for EMA systems are available here
.
Please note that MFA is not acvated in the CTIS Training Environment.
2.2. Organisaon and Sponsor Administrator registraon
2.2.1. Organisaon registraon in OMS
CTIS ulises organisaon data from Organisaon Management Service (OMS). OMS provides a single
source of validated organisaon data that can be used as a reference to support EU regulatory acvies
and business processes. It stores master data comprising organisaon name and locaon address for
organisaons such as markeng authorisaon holders, sponsors, regulatory authories, trial sites and
manufacturers.
If an organisaon has already been successfully registered in OMS, its informaon is public and a user can
search and retrieve its details within CTIS to populate the CTA, submit noficaons, or to use it for other
sponsor-related acvies in CTIS (e.g. populate employer’s details in personal profile).
The possibility to search and retrieve an organisaon in CTIS is available in different areas across CTIS,
and these areas are provided in the following table. Users are recommended to register in OMS the
organisaons in advance to the populaon of their inial CTA.
Table 2.2.1.1. Search and retrieval of an organisaon from OMS in CTIS.
CTIS Locaon/Situaon
OMS data search & retrieval
(through pop-up windows)
Personal profile Update employer informaon
Request a role Add sponsor organisaon
Create new trial Add sponsor organisaon
Part I: ‘Trial details’ ‘Scienfic advice and Paediatric
Invesgaon Plan (PIP)’ secon
Add a Competent Authority
Part I: ‘Sponsor’ secon Add Sponsor Legal contact / Add Third party organisaon
Part II: Trial Sites Add site
Serious Breach noficaon form Add site (where the breach occurred)

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 12/62
If an organisaon is not yet registered in OMS when starng to use CTIS, it is required first to register the
organisaon via a ‘change request’
1
directly on the OMS portal. OMS can register organisaons included
in a Naonal Business Registry.
It is paramount that requests are supported by valid documentaon to achieve a successful OMS
validaon. Therefore, it is expected that the enty in possession of the valid documentaon proceeds
with the registraon in OMS. Users are urged to access the OMS related training material stored in
OMS
portal document repository and view the document requirements to submit a change request found in
the document ‘E - OMS Change Requests’.
Sponsor organisaons that are not registered in any Naonal Business Registry and cannot provide EMA
with sufficient documentaon for their requests as per document ‘E - OMS Change Requests’, shall aach
to their requests a ‘CT registraon Headed leer’ found as a template in
OMS portal document
repository.
In case the request raised is incorrect, or it is not supported by the appropriate accompanying
documentaon, this will result in the OMS validaon failing, and the organisaon’s details will not be
retrievable in a subsequent search in OMS for that organisaon.
Of note, any party can create a new organisaon record in OMS e.g., for a clinical invesgator site, CROs,
vendors or other facilies etc., that would be required when compleng a CTA or subming a
otherwise it will not be possible for the sponsor user to search, select and find that organisaon’s details
again if the OMS request has failed (e.g. to support the registraon in OMS of a clinical invesgator
site/facility, a headed leer document signed and dated by a representave of that organisaon, stang
the full company name and address, should be provided).
Sponsors should refer to OMS process to ascertain the validaon melines of change requests to be able
to search and select the organisaon with the address of interest in CTIS. Note that an organisaon can
have several addresses (linked to the same main ORG-ID) and only one can be selected in CTIS.
References
Locaon (area or document)
EMA Organisaon Management Service
(OMS) homepage
hps://www.ema.europa.eu/en/human-
regulatory/research-development/data-medicines-iso-
idmp-standards/spor-master-data/organisaon-
management-service-oms
Submission of change requests in OMS
hps://www.ema.europa.eu/en/human-
regulatory/research-development/data-medicines-iso-
idmp-standards/spor-master-data/organisaon-
management-service-oms#subming-change-
requests-secon
Industry Webinar Introducon to OMS
services and acvies
hps://www.youtube.com/watch?v=fxMpsgDnWZY&ab
1
The term ‘change request’ for OMS refers to an addion of new or modificaon of exisng records in OMS.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 13/62
2.2.2. Organisaon registraon locally in CTIS for use in CTIS
If users do not retrieve an organisaon further to a search in OMS or in CTIS, they can opt to create the
site in CTIS by clicking the buon ‘New Organisaon’, which will now appear enabled. It should be noted
that this funconality is only available for:
• Part I Sponsor secon – Third-party organisaons
• Part II – Trial sites
• Serious Breach noficaons – Details of the site where the serious breach occurred
• Third Country Inspectorate Inspecon – Third country inspecon site
• MS Inspecons – Inspected site
Organisaons that are created locally in CTIS are not validated by EMA. But users are encouraged to keep
in mind the OMS data quality standard set when they create their organisaons in CTIS, reducing then
the possibility to receive RFIs from assessing authories regarding inaccurate data.
Once users iniate the registraon of an organisaon in CTIS, it is in DRAFT status and the dra
organisaon is visible only within the scope of the dra CTA or dra noficaon, i.e. it does appear when
other sponsors (or even the same sponsors who created the organisaon) search in CTIS.
Once the CTA or noficaon, which contains this organisaon registered in CTIS (sll in DRAFT status) is
submied, the locally registered organisaon in CTIS changes from DRAFT status to ACTIVE status. This
implies that the organisaon is now searchable by other users, including users from different
organisaons such as other sponsors. Organisaons registered locally in CTIS with ACTIVE status are no
longer editable (to edit details of the organisaon such as address or name or country etc). If users need
to update the organisaon details whilst responding to an RFI or draing a substanal modificaon, they
will need to remove the inially created organisaon from their applicaon/noficaon and add a new
More details can be found in the Module 03 of the CTIS Training Material Catalogue in
Step-by-step guide
(Create organisaons locally in CTIS).
2.2.3. Sponsor Administrator registraon in EMA Account Management portal for use in CTIS
The CTIS Sponsor Administrator is a high-level administrator role requested and managed through the
EMA Account Management portal.
A sponsor administrator is required to iniate the management of users in the sponsor workspace.
The request for the Sponsor Administrator role is submied by the user that will become the Sponsor
Administrator for an organisaon with a specific organisaon idenfier (ORG-ID) and will be handled via
EMA Account Management portal.
The registraon process for the Sponsor Administrator (‘Sponsor Admin’) role, via the EMA Account
Management portal, started on 1 September 2021 and needs to be supported by an appropriate
‘Affiliaon leer’ submied to EMA at the me of registraon. There is also the alternave route of the

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 14/62
External Organisaon Administrator, described in the EMA Account Management portal (last link in
‘References’ table below).
It should be noted that the appointment of the high-level administrator for a sponsor, namely the
Sponsor Admin for an organisaon, is processed in IAM based on the organisaon idenfier (Org-ID).
If different Org-IDs for the same organisaon exist, they cannot be grouped together in the request to
appoint a Sponsor Admin for the purpose of using CTIS, and therefore, each request should be raised
individually. However, the same affiliaon leer can be used and aached for each individual request.
Once the Sponsor Administrator role is assigned to one person by EMA (or by the External Organisaon
Administrator of the organisaon) on the basis of the validaon of the request, the Sponsor
Administrator (or the External Organisaon Admin) receives automated e-mail noficaons of requests
from other users wishing to become Sponsor Administrators for the same organisaon. The first Sponsor
Admin (or the External Organisaon Admin) manages such requests in the EMA account Management
portal.
EMA does not handle these requests once the first Sponsor Administrator (or an External Organisaon
Administrator) has been assigned by EMA. The necessity of an ‘Affiliaon leer’, or any other supporng
documentaon, for these subsequent requests are decided by each organisaon internally
2
.
Explanatory training material is available in the CTIS Training Material Catalogue, in Module 07
‘Management of registered users and Role matrix’ and on the EMA Account Management homepage.
References
Locaon (area or document)
EMA Account Management homepage
hps://register.ema.europa.eu/identyiq/home.ht
ml
Affiliaon leer template
hps://register.ema.europa.eu/identyiq/help/affili
aon template.docx
CTIS Training Material Module 07 ‘Management
of registered users and Role matrix: Step-by-
step Quick Guide (high level Admin)’
hps://www.ema.europa.eu/en/documents/other/
step-step-guide-high-level-cs-administrator-
management-roles-permissions-cs-training-
programme_en.pdf
CTIS Training Material Module 07 ‘Management
of registered users and Role matrix: Frequently
Asked Quesons (FAQs) – specific ones’
hps://www.ema.europa.eu/en/documents/other/
faqs-management-roles-permissions-cs-training-
programme-module-07_en.pdf
CTIS Training Material Module 03 ‘Frequently
Asked Quesons (FAQs)’
hps://www.ema.europa.eu/documents/other/faq
s-user-access-management-cs-training-
programme-module-03_en.pdf
CTIS Training Material Module 07 ‘How to
request roles and how to assign roles to
registered users in CTIS’ and ‘How to amend
hps://www.youtube.com/watch?v=CBLVMFC4JeA
hps://www.youtube.com/watch?v=1CUyQcICyl8
2
However, proof of affiliaon leer is not required once an External Organisaon Administrator has been validated in EMA Account Management for a
certain organisaon.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 15/62
and revoke roles of registered users in CTIS’
Videoclips
External Organisaon Administrator
hps://register.ema.europa.eu/identyiq/help/user
admin.html#OrganisaonAdmin
3. Management of users and organisaons in CTIS
3.1. Key user management concepts in CTIS
There are two approaches to user management in CTIS: the organisaon-centric approach and the trial-
centric approach.
These approaches have been designed according to the needs of the different types of sponsor
organisaons that will use CTIS.
Before using CTIS, sponsors should carefully consider which user management approach best fits their
organisaon.
A full descripon of each of these approaches, including their advantages and disadvantages, are
explained in the reference documents listed below, as well as in Secon 3.3 (‘Organisaon-centric
approach - Sponsor Administrator’) and secon 3.4. (‘Trial-centric approach – Clinical Trial
Administrator’) of this document. Based upon the current experience with user administraon for non-
commercial sponsors, we recommend considering following the organisaon centric approach and to
request the help of your Clinical Trial Centre for CTIS operaons and CTR compliance.
References
Locaon (area or document)
CTIS Training Material Module 07 ‘Creang a
clinical trial: Clinical trial centric approach vs
organisaon centric approach’ (Video)
hps://www.youtube.com/watch?v=hfzZxwX2W-Y
CTIS Training Material Module 07 ‘Management
of registered users and role matrix’
hps://www.ema.europa.eu/en/learning-
module/management-roles/story.html
3.2. User roles concept in CTIS
In order to perform an acon in CTIS, such as preparing, subming or viewing a CTA, noficaons,
summary of results or clinical study reports, a user must be assigned with a CTIS user role to obtain
appropriate permissions.
Up to 18 sponsor user roles are foreseen for CTIS. The profile of a user can be built with a combinaon of
different roles, to allow the user to complete various acons in CTIS. Users with administrator roles (high-
level administrator, clinical trial administrator) can assign roles to other users, enabling them to perform
acons.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 16/62
Each role in CTIS comes with a specific set of permissions, which are predefined levels of acons that
users can perform on data and documents stored in CTIS. These permissions are at user management
level (reserved for administrator user roles) and access level, ranging from viewing to creang, preparing
EMA has prepared a document to describe the concept of user roles and permissions in detail, a Role
Matrix, which outlines the permissions linked to each user role, and a summary of roles document. These
documents can be found at the links below.
References
Locaon (area or document)
CTIS Training Material Module 07
‘Management of registered users and role
matrix’
hps://www.ema.europa.eu/en/learning-
module/management-roles/story.html
Sponsors Business Processes and Roles
hps://www.ema.europa.eu/documents/other/sponsors-
business-processes-roles-cs-training-programme-
module-07_en.pdf
Sponsor workspace: summary of role
permissions - Sponsor Workspace Roles -
permission matrix summary
hps://www.ema.europa.eu/documents/other/roles-
permissions-matrix-summary-sponsors-workspace-cs-
training-programme-module-07_en.pdf
3.3. Organisaon-centric approach - Sponsor Administrator
The organisaon-centric approach is one of two user management approaches in CTIS that can be used
by sponsors of a clinical trial. It is intended to serve the needs of organisaons and/or sponsors that run
mulple clinical trials.
The organisaon-centric approach means that user management is done at organisaon level.
Under the organisaon-centric approach, the sponsor needs to appoint a high-level administrator
(Sponsor Administrator). The Sponsor Administrator must be registered in EMA Account Management
plaorm (see Secon 2.2.2 on ‘Sponsor Administrator registraon in EMA Account Management portal
for use in CTIS’).
Before a user can register as a high-level administrator for a sponsor organisaon, this organisaon needs
to be registered with the Organisaon Management Service (OMS); see Secon 2.2.1 on ‘Organisaon
registraon in OMS for use in CTIS’.
Management of users within the organisaon is done at the organisaon level with a top-down model.
Once appointed, sponsor administrators can assign medium-level administrator (i.e. clinical trial
administrator) and business roles to users in CTIS to perform user management or business acvies. In
the organisaon-centric approach, users become affiliated to the organisaon (in parcular, the user
becomes affiliated to the ORG-ID number as registered in OMS) of the sponsor administrator in CTIS
when they are assigned with a role by this administrator.
The organisaon-centric approach is parcularly useful for organisaons that (will) conduct trials on a
regular basis, even if the frequency is low. The advantages of this approach are that it allows the

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 17/62
management of access and roles across trials within one organisaon thus, supporng data quality and
integrity through a top-down validaon process, as well as ensuring security as a user can only create a
new CTA for that organisaon-ID, as registered in OMS, if it has been previously assigned the clinical trial
administrator (CT Administrator) role by the sponsor administrator.
Note: a user needs to be given the role of CT Administrator with scope ‘all trials’ in order to be able to
create one or more new CTAs, copy or resubmit a trial for that organisaon-ID.
Addional informaon is published on the EMA Corporate website under the Training Programme - User
access management (Module 03).
References
Locaon (area or document)
EMA Account Management homepage
hps://register.ema.europa.eu/identyiq/home.html
CTIS Training Material Module 03 ‘User
access management: User Quick Guide’
hps://www.ema.europa.eu/en/documents/other/quick-
guide-user-access-management-cs-training-programme-
module-03_en.pdf
3.4. Trial-centric approach – Clinical Trial Administrator
The trial-centric approach is one of two user management approaches in CTIS. A user is automacally
guided to use this approach in CTIS only in the case a sponsor administrator has not been registered and
appointed in the EMA account management system for a specific organisaon.
In this approach, when the user iniates the creaon of a new CTA, the system checks if a sponsor
administrator has been appointed for the sponsor organisaon selected for that inial CTA . If that is not
the case, the user will be able to proceed becoming the clinical trial administrator for that parcular trial.
Further allocaon of other CT Administrator or business roles to users is then done at trial level. The
clinical trial administrator can manage users only for the trial(s) of his/her concern and can perform all
sponsor business acvies in CTIS related only to that parcular trial(s).
In the trial-centric approach, users follow a boom-up model that supports an easy way of subming a
limited number of CTAs and straighorward management of a small number of users at trial level, not
organisaon level.
This approach is intended to serve the needs of small organisaons and specifically academic sponsors,
which may iniate trials on an ad hoc basis. It allows for the management of a smaller number of users
and one or very limited numbers of clinical trials. This allows a faster process (no need for registraon of
a high-level sponsor administrator) when subming a first inial, and subsequent applicaons, as
applicable. However, it is less secure as any user can, potenally, create a trial on behalf of a sponsor
organizaon that has not previously registered a sponsor administrator. Moreover, no individual user will
have a centralised oversight of the trials being conducted for that sponsor organizaon nor the users
involved.
Addional informaon is published on the EMA corporate website under the Training Programme - User
access management (Module 03).

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 18/62
References
Locaon (area or document)
CTIS Training Material Module 03 ‘User access
management: User Quick Guide’
hps://www.ema.europa.eu/en/documents/other/q
uick-guide-user-access-management-cs-training-
programme-module-03_en.pdf
3.5. Markeng Authorisaon Holder (MAH) group of users
A MAH Administrator role is also available to support the submission of clinical study reports into CTIS
when a trial has been included in a markeng authorisaon applicaon. The registraon process for MAH
Administrator takes place via CTIS Service Desk, with the submission of a valid cover leer, including the
required informaon. More details on this process can be found in the CTIS Training Material Module 13
‘Clinical study reports submissions’.
References
Locaon (area or document)
CTIS Training Material Module 13 ‘Clinical
study reports submissions’
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-cs-online-
modular-training-programme#sponsor-workspace-
secon
3.6. CTIS user personas and organisaon models
Sponsors have various processes, structures and partnerships for managing clinical trials. In order to
facilitate the compleon of processes in CTIS, sponsors must understand the CTIS User Management
funconalies and how to best make use of these funconalies in their organisaonal environment.
Sponsors also need to understand user roles so that they can ensure that they have the correct
confidenality agreements in place.
To assist sponsors in understanding the CTIS User Management funconalies and how to organise in
CTIS, EMA has published CTIS user personas linked to CTIS user roles and permissions, and example
sponsor organisaon models in CTIS.
References
Locaon (area or document)
CTIS sponsor user personas
hps://www.ema.europa.eu/documents/other/clinic
al-trial-informaon-system-cs-sponsor-user-
personas_en.pdf
Principles for sponsor organisaon modelling
in CTIS
hps://www.ema.europa.eu/documents/other/princ
iples-sponsor-organisaon-modelling-cs_en.pdf

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 19/62
4. How to get a clinical trial applicaon started in CTIS
4.1. Introducon
Under the organisaon-centric approach, the Sponsor Administrator needs to organise access for the
users based on the interacons that they will have in the system. It is essenal that the permissions
granted, and the scope of the role (trial specific or all trials) are understood to enable the relevant
navigaon, access to data and documents accordingly but also to enable acvies in the system as
required. Refer to Secons 2. and 3.
In case, the trial centric approach is selected, the CT administrator needs to organise user access based
on the interacons users should have in the system to fulfil the sponsor’s obligaons.
It is important that in advance of any interacons with the system, sponsor users get familiarised with the
use and navigaon in CTIS. It is recommended that inially a small team of users is assigned to navigate
to perform acvies in CTIS for a given trial/applicaon. These users are strongly recommended to get
experience first on the CTIS Training environment (see Secon 10.5. ).
This approach enables familiarisaon with CTIS and facilitates readiness and preparaon of dossier to
ease member state assessment.
To create and prepare any type of clinical trial applicaons in CTIS, several training materials including
step-by-step, e-learning and videos can be consulted (module 10). Secon 4.2 presents the various
subsecons of the clinical trial applicaon tab as they need to be completed and their corresponding
dedicated training materials. An overview of the documents available to be submied in CTIS is also
presented. Secon 4.3 presents the navigaon of the evaluaon tab and the melines that provide
informaon on the assessment acvies once the clinical trial applicaon has been submied. All
melines due dates in CTIS follow Central European Time (CET), regardless of the seasons of the year.
4.2. Draing of the clinical trial applicaon dossier
Table 4.2.1. Create a CTA
Step process 1: Create a CTA
Locaon (area or document)
Create a clinical trial applicaon (Step-by-
step guide)
hps://www.ema.europa.eu/en/documents/other/ste
p-step-guide-create-submit-withdraw-clinical-trial-
applicaon-nonsubstanal-modificaons-cs_en.pdf
Create a clinical trial applicaon (E-learning)
hps://www.ema.europa.eu/en/learning-
module/create-ct-applicaon/story.html
Table 4.2.2. Populate MSC & Form secons
Step process 2: Populate MSC & Form secons
Locaon (area or document)
Complete the MSC and Form secons (video)
hps://www.youtube.com/watch?v=1du3VUq4K5g

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 20/62
Table 4.2.3. Populate Part I secon
Step Process 3: Populate Part I secon
Locaon (area or document)
Complete the Part I secon (video)
hps://www.youtube.com/watch?v=piRl9ZGTe-Y
Fill in the trial details of Part I secon (video)
hps://www.youtube.com/watch?v=q2Qn6p9VnXs
Complete sub secon Sponsor details (video)
hps://www.youtube.com/watch?v=4HtR_Xtn7pc
Complete sub secon Product details (video)
hps://www.youtube.com/watch?v=e-JTvFoBlCs
Sponsor Handbook v.3.0.3: Secon 6 ‘Product
management in CTIS’
Table 4.2.4. Populate Part II secon
Step Process 4: Populate Part II secon
Locaon (area or document)
Complete Part II secon (video)
hps://www.youtube.com/watch?v=jmylMwZFroc

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 21/62
Table 4.2.5. Overview of documents and data available to be submied in CTIS for an inial CTA.
CTA tabs
subsecons
CTA placeholders
Document types available
for upload
3
,
4
,
5
Note
Form
Cover leer
Cover leer
Refer to the EudraLex
vol.10 CTR Q&As and CTR
document on content
requirements
Proof of payment
Proof of payment
Where applicable
Compliance with regulaon
Compliance with Regulaon
(EU) 2016/679
Trial category
Complete the relevant data
field, see secon 7.2.
MSC
Member states concerned
Indicate the proposed RMS
Part I
Trial specific informaon
(Part I) – Trial details
Trial idenfiers
Provide idenfiers of the
trial
Trial informaon
Low Intervenon
jusficaon
Protocol informaon
• Protocol
• Protocol synopsis
• DSMB
• Study design
Scienfic advice and
Paediatric Invesgaon Plan
(PIP)
•
Summary of scienfic
advice
• Scienfic advice–Quality
• PIP opinion
Associated clinical trials
Sponsor agreement
3
Include a document ‘not for publicaon’ alongside the document ‘for publicaon’ is needed.
4
Include translaons if required; refer to the CTR Q&As (Eudralex vol.10 chapter V)
5
Only include signed documents if required as per the CTR Q&As (Eudralex vol.10 chapter V)

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 22/62
CTA tabs
subsecons
CTA placeholders
Document types available
for upload
3
,
4
,
5
Note
References
Provide the PMID
6
number
if available
Countries outside the EEA
Add non-EEA country if
applicable
Trial specific informaon
(Part I) – Sponsor details
Refer to the Eudralex
vol.10 CTR Q&As on
content requirements
Trial specific informaon
(Part I) – Product details
Reference to content (CTA)
Role: test
• IB or SmPC
• IMPD safety and efficacy
• IMPD Quality
Include comparator,
placebo or auxiliary
product as necessary
Content labelling
Content labelling of IMP
Part II
Trial site
Recruitment arrangements
Recruitment arrangements
Refer to ‘Part II applicaon
document templates’,
available under chapter I of
Eudralex vol.10
Subject informaon and
informed consent form
Subject informaon and
informed consent form
Refer to naonal
requirements
Suitability of the
invesgator
• Invesgator CV
• Suitability of the
invesgator
• Declaraon of Interest
Suitability of the facilies
Suitability of the facilies
If signed, consider
redacon
6
PubMed Idenfier

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 23/62
CTA tabs
subsecons
CTA placeholders
Document types available
for upload
3
,
4
,
5
Note
Proof of insurance cover or
indemnificaon
Proof of insurance cover or
indemnificaon
Only the cerficate of
insurance (avoid adding
CCI in these documents)
Financial and other
arrangements
Financial and other
arrangements
Naonal requirements, e.g.
some MS require signed
Clinical Trial agreements
Compliance with naonal
requirements on Data
Protecon
Compliance with naonal
requirements on Data
Protecon
Oponal only, required by
some countries
Compliance with use of
Biological samples
Compliance with use of
Biological samples
Oponal, only if biological
samples are collected
References
Locaon (area or document)
Technical requirements for opmal use of CTIS
hps://www.ema.europa.eu/en/documents/other/
clinical-trials-informaon-system-cs-technical-
requirements-opmal-use_en.pdf
CTIS training material module 10 ‘Create,
submit, and withdraw a clinical trial'
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-cs-online-
modular-training-programme#sponsor-workspace-
secon
Addional reference materials for CTIS users
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-training-
support#addional-reference-materials-for-cs-
users-secon
CTIS Structured data form Instrucons - inial
applicaon, addional MSC, substanal and
non-substanal modificaons
hps://www.ema.europa.eu/documents/template-
form/clinical-trial-informaon-system-cs-
structured-data-form-inial-applicaon-addional-
member_en.xlsx
List of data and documents requested as a
minimum in CTIS to proceed with submission of
the different applicaon types
hps://www.ema.europa.eu/en/documents/other/
checklist-required-fields-applicaon-type-cs-
training-programme-module-10_en.pdf

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 24/62
4.3. Clinical Trial Applicaon under evaluaon
Table 4.3.1. Evaluaon Tab
The Evaluaon tab includes several subsecons presenng the assessment overview for the validaon,
the Part I and the Part II, and the decision for each MSC. The validaon, Part I, and Part II secons include
an RFI and a conclusion subsecon.
Evaluaon tab
Locaon (area or document)
View the tabs for validaon, Part I conclusion,
Part II conclusion, and decision of a clinical
trial applicaon
Search, view, and download informaon on a
clinical trial
hps://www.ema.europa.eu/en/documents/other/q
uick-guide-how-search-view-download-clinical-trial-
clinical-trial-applicaon-sponsors-cs_en.pdf
Access, view, and respond to an RFI
hps://www.youtube.com/watch?v=vbQVkYi3pGI
The metables give an overview and progress on the assessment of the parcular clinical trial
applicaon.
Table 4.3.2. Timetables and Timelines for CTA Assessment
CTA Assessment: Timetables and Timelines
Locaon (area or document)
View Timetable (video)
hps://www.youtube.com/watch?v=HN7zcQW81P0
CTIS Evaluaon melines
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-training-
support#evaluaon-melines-secon
4.4. Clinical Trial Applicaon aer decision
Once the clinical trial applicaon has been authorised, the conclusion to the validaon and the
assessment phases can be viewed in the evaluaon tab. At the boom of the page, in the assessment
overview table, the informaon is recorded for each member state and includes each MSC’s respecve
decision.
In the event that a MSC would have disagreed to the conclusion ‘acceptable’ or ‘acceptable with
condions’ to Part I, this informaon would be also recorded in that secon.
All documents and data authorised for the trial, as a result of the authorisaon of the latest applicaon,
can be consulted in the tab ‘Full Trial Informaon’.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 25/62
5. Transion from Direcve to Clinical Trial Regulaon
5.1. Transion period
There is a 3-year transion period that started on the CTIS go-live date.
Year 1 (31 January 2022 to 30 January 2023):
During the first year aer CTIS go-live, sponsors could choose whether to apply for a new CTA under the
Clinical Trial Direcve (CTD: Direcve 2001/20/EC), or to apply under the new legislaon, the Clinical Trial
Regulaon (EU) No 536/2014, using CTIS.
Member States were ready to use CTIS and accept applicaons under the new legislaon (CTR) from day
1 of CTIS go-live.
Years 2 and 3 (31 January 2023 to 30 January 2025):
From 31 January 2023, all new CTAs must be submied under the new legislaon (CTR) using CTIS.
Submission of new CTAs under the CTD in EudraCT is no longer be available for new CTAs. The addion of
new member states is also no longer be possible under the CTD as from 31 January 2023. Trials under the
CTD must first be transioned, and then an Addional Member Stated Concerned Applicaon can be
submied only once the clinical trial dossier has been updated through a substanal modificaon using
CTIS.
Figure 5.1.1. Schemac representaon of the Clinical Trial Regulaon transion period.
CTAs that were submied under the old legislaon (CTD), ulising EudraCT, prior to 30 January 2023, will
be able to connue to run unl compleon under that Direcve unl 30 January 2025. Processes will
remain unchanged, and sponsors will therefore be able to submit substanal amendments and end of
trial noficaons as needed under the Direcve. EudraCT will remain operaonal throughout the
transion period to enable these trials to connue.
As from 31 January 2025, clinical trials authorised under the CTD must either have ended in the EU/EEA or
have been transioned. They cannot connue operang under the old legislaon ulising EudraCT beyond
the end of the 3-year transion period (30 January 2025). Thus, if sponsors are running trials that they
expect to connue in EU/EEA beyond 30 January 2025, sponsors need to transion them to the CTR before
the transion period expires.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 26/62
Figure 5.1.2. Schemac representaon of the Clinical Trial Regulaon transion period from 31 January 2023.
EudraCT will remain acve beyond the end of the transion period for sponsors to nofy global end of the
trial and submission of summary results of trials completed under the Direcve.
Transion applicaons can be submied at any me during the 3-year transion period and sponsors are
urged to ensure that they complete the process early enough in the transion period to ensure connuity
of the clinical trial beyond 30 January 2025, taking account of statutory holidays and the two-week winter
clock stop.
References
Locaon (area or document)
Link to EudraLex - Volume 10 - Clinical trials
guidelines - Set of documents applicable to
clinical trials authorised under Regulaon EU No
536/2014
7
hps://ec.europa.eu/health/documents/eudral
ex/vol-10_en#fragment1
Guidance for the Transion of clinical trials from
the Clinical Trials Direcve to the Clinical Trials
Regulaon
hps://health.ec.europa.eu/document/downlo
ad/10c83e6b-2587-420d-9204-
d49c2f75f476_en?filename=transion_ct_dir-
reg_guidance_en.pdf
CTCG Best Pracce Guide for sponsors of
mulnaonal clinical trials with different Part I
document versions approved in different Member
States under the Direcve 2001/20/EC that will
transion to the Regulaon (EU) No. 536/2014
hps://www.hma.eu/fileadmin/dateien/HMA_j
oint/00-_About_HMA/03-
Working_Groups/CTCG/2024_03_CTCG_Best_Pr
acce_Guide_for_sponsors.pdf
Annex Cover Leer Template Declaraon vs. 4.0
hps://www.hma.eu/fileadmin/dateien/HMA_j
oint/00-_About_HMA/03-
Working_Groups/CTCG/2024_03_CTCG_Annex_
cover_leer_template_-
7
The latest published European Commission Clinical Trials Regulaon No 536/2014 Q&A document can be found under the ‘Set of documents applicable
to clinical trials authorised under Regulaon EU No 536/2014’ secon.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 27/62
_CTCG_Best_Pracce_Guide_for_sponsors_on_
transion_vs4.docx
5.2. Points to consider on transional arrangements
Some aspects have been included here of what the sponsor should consider when defining a submission
strategy for CTIS during the transion period.
5.2.1. What trials should not be transioned
• Trials that have already ended or will end before the end of the transion period in the EU/EEA
should not be transioned.
• If an end of trial noficaon has been submied in all EU/EEA member states, but the global end of
the trial has not been nofied, the trial should not need to be transioned. Global end of the trial
and trial summary results should be posted via EudraCT under the Direcve.
• Trials that are old and started prior to the Direcve 2001/20/EC coming into applicaon do not
benefit from the transion process. If they are truly intervenonal and need to connue to run aer
the end of the CTR transion period, then a new CTA under the CTR needs to be submied.
• Paediatric trials that are being conducted enrely outside the EU/EEA but for which a EudraCT
number has been created should also not be transioned.
5.2.2. Can the trial be transioned?
Only trials submied under the CTD and likely to be ongoing beyond 30 January 2025 need to be
transioned if they meet these criteria:
• are intervenonal clinical trials in humans;
• involve at least one acve site in the EU/EEA where the trial is sll ongoing;
• there are no substanal amendments ongoing in any Member State Concerned (MSC) under the
Direcve.
Details of the requirements for transioning of mono-naonal and mul-naonal trials are provided in
the EudraLex Volume 10 Q&A menoned in the References table above.
General consideraons:
Sponsors need to ensure that all current approved documents under the Direcve are available in
electronic format in compliance with CTIS upload requirements (see secon 7.1.3. ‘Data fields and
document specificaons’).
Retrospecve documents do not need to be submied to CTIS (e.g. earlier versions of IBs or Protocols
that have been superseded under the CTD). Only current approved versions should be included in the
transion applicaon.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 28/62
Where a mandatory document is expected to be uploaded into the CTIS that does not exist for the
transioning trial (e.g. site suitability documentaon), then a blank document is expected to be uploaded
with a comment that the document does not apply and it has been provided to allow transion from the
CTD to the CTR Regulaon.
When compleng the CTA, and providing the CT data and documents in CTIS, consideraon should be
given to the transparency requirements of the CTR, including the need to remove personal data from
submied documentaon, if applicable: see chapter 8. on transparency of data.
Mono-naonal trials:
The trial is transioned from the CTD to the CTR by subming a new applicaon in CTIS that reflects the
content of the dossier that is currently approved and has been assessed by the MSC. Documentaon
required is specified in the queson 9 to the
Guidance for the Transion of clinical trials from the Clinical Trials
Direcve to the Clinical Trials Regulaon the available in the EudraLex Volume 10
Mul-naonal trials:
Mul-naonal clinical trials (trials conducted under the same EudraCT number in different Member
States) should be transioned as a single mul-country CTA under the CTR, ulising a harmonised or at
least consolidated protocol Sponsors may need to consider harmonising the protocol by substanal
amendments under the CTD before they transion them as one trial under CTR with one EU Clinical Trial
number Consolidaon of a protocol, to reflect only what is already approved in each MSC prior to
submission of a transion applicaon, does not require prior approval via submission of a substanal
amendment under the Direcve since no changes to the protocol content are made during the
consolidaon process.
In addion, if a sponsor intends to transion a mul-naonal clinical trial as a mul-country CTA under
the CTR, only data field informaon and documents for the MSCs where the trial is sll ongoing need to
be entered in CTIS.
The trial is transioned from the Direcve to the Regulaon by subming a new applicaon in CTIS that
reflects the dossier that is currently approved and has been assessed by all the MSCs. If the protocol is
not consolidated, the sponsor should first submit a substanal amendment under the Direcve in order
to align and obtain a harmonised protocol authorised by all Member States before subming a transion
applicaon under the CTR.
Alternavely, for trials where full harmonizaon of the protocol to be submied in the Part I of the
applicaon cannot be achieved due to different naonal requirements, a sponsor needs to prepare a
consolidated protocol, reflecng the common core provisions and capturing the minor differences as
regards the naonally authorised trials (see Reference table above for CTCG’s ‘Best Pracce Guide for
sponsors of mulnaonal clinical trials’). The consolidated protocol must correspond to what is
authorised in each of the Member States concerned.
For VHP trials being transioned, the sponsor should propose as the RMS the country that acted as the
VHP Reference MS.
For more informaon regarding the condions for the transion of mul-naonal trials refer to the
queson 10 to the Guidance for the Transion of clinical trials from the Clinical Trials Direcve to the
Clinical Trials Regulaon the available in EudraLex Volume 10.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 29/62
5.2.3. What are the assessment melines of transional trials
In CTIS aer submission of the dossier, the assessment workflow is triggered and the MSC need to select
the RMS in the event of mul-naonal trials. The validaon, assessment Part I, Part II and decision
milestones are triggered in the system with the need for MSC to proacvely record their conclusions and
decisions. Therefore, transioning a trial from the Direcve to the Regulaon can take up to a period of
60 days.
It is unlikely that RFI are raised for transional trials unless the documentaon submied does not
correspond with the documents approved under the CTD. In such a case, the melines would be
extended by 15 days in the validaon or 31 days in the assessment phase.
5.3. How to create a transional trial in CTIS
To transion a trial from EudraCT to CTIS, sponsors need to submit an inial CTA marked as a transional
trial. On how to mark their inial CTA as transional trial, users may follow the instrucons of the Quick
Guide of CTIS Training Module 23 that is dedicated to transional trials. Once an inial trial is marked as
transional, addional fields will appear in the applicaon dossier, allowing users to add in their
transional trial the corresponding EudraCT trial number. If sponsors create the trial without marking it
as transional, they cannot correct it at a later stage. They shall create the trial from scratch, marking it
this me as transional.
References
Locaon (area or document)
CTIS Training Material Module 23 –
Transional trials: ‘Quick guide Transional
trials from EudraCT to CTIS’
hps://www.ema.europa.eu/en/documents/other/s
ponsors-guide-transion-trials-eudract-cs-cs-
training-programme-module-23_en.pdf
CTIS Training Material Module 23 –
Transional trials: ‘FAQs Transional trials
from EudraCT to CTIS’
hps://www.ema.europa.eu/en/documents/other/fa
qs-transion-trials-eudract-cs-cs-training-
programme-module-23_en.pdf
5.4. How to manage trials transioned to the CTR in CTIS
Once the trial has a recorded authorisaon in CTIS, all the requirements of the CTR apply from the date
of approval of the transion applicaon under the CTR. The sponsor needs to comply with their CTR
obligaons for the management of the trial and submit noficaon informaon as required. These
include start of trial noficaon and start of recruitment that can have occurred prior to authorisaon,
but could include further events as they are likely to take place.
Also, any changes to the dossier need to be reflected in line with the requirements of the CTR. Therefore,
any subsequent substanal modificaons submied to the MSC need to comply with the requirements of
the CTR.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 30/62
6. Product management in CTIS
6.1. Medicinal product registraon in XEVMPD
Before compleng the CTA in CTIS, the sponsors should ensure that the details of the medicinal products
used in the clinical trials are already registered in the eXtended EudraVigilance Medicinal Product
Diconary (XEVMPD). It should be noted that a placebo can be added manually in CTIS directly; sponsors
do not need to submit placebo’s informaon on the XEVMPD.
The diconary includes all medicinal products that are authorised in the EU/EEA and unauthorised
medicinal products (referred to in the XEVMPD as 'development' products) that are associated with
clinical trials. Unauthorised products include those that have not received a markeng authorisaon in
the EU/EEA for the strength and/or pharmaceucal form.
To submit medicinal product data in the XEVMPD, sponsor organisaons must be registered in the
Organisaon Management Service (OMS) and also with EudraVigilance either via Gateway or the
EudraVigilance web applicaon (EVWEB). This applicaon allows registered users to create and send
Extended EudraVigilance Product Report Messages (XEVPRMs), receive XEVPRM acknowledgements,
view medicinal product informaon and perform queries.
Consolidated guidance on the electronic submission of informaon on unauthorised medicinal products
for human use in the XEVMPD is now available on the
'Data submission on invesgaonal medicines:
guidance for clinical trial sponsors' webpage. The guidance was prepared based on the processes already
in use and on informaon available in exisng documentaon.
Some high-level details specific for the registraon of medicinal products in XEVMPD, to then be used in
CTIS, are also presented below to describe the business flow.
The acve substance for the development medicinal product must be available in EMA SMS (Substance
Management Service).
Substance data is entered and maintained in the XEVMPD by the EMA; when substance informaon is
successfully inserted in the XEVMPD, a substance EV Code is generated by the XEVMPD.
To request the addion of new substance informaon, or an amendment of exisng substance
informaon, in the XEVMPD, sponsors should follow the process described in the
'Changes to some
business rules of the eXtended EudraVigilance Medicinal Product Diconary (XEVMPD)' document. EMA
will validate the request and the substance EV Code will be provided to the sponsor via an e-mail
confirmaon from the
EMA ServiceNow within 4 working days.
If a development medicinal product needs to be entered in the diconary by the sponsor, the sponsor
should submit the medicinal product data in the XEVMPD via an XEVPRM with the operaon type 'Insert'.
The medicinal product data must be submied in accordance with the principles described in secon 1
‘Inial submission of a development medicinal product’ of the
'Guidance on the electronic submission of
informaon on invesgaonal medicinal products for human use in the Extended EudraVigilance
medicinal product diconary (XEVMPD)' document. The document also includes informaon on how to
add missing informaon (for example substance or sponsor details) in the XEVMPD. Providing that the
inseron was successful, an EV Code will be assigned to the medicinal product record by the XEVMPD
and sent automacally to the sponsors' sender organisaon ID via an XEVPRM acknowledgement.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 31/62
Once the EV Code assigned to the medicinal product record is available in the XEVMPD, the sponsor can
search and retrieve the product details in CTIS. More informaon on associang an unauthorised
medicinal product to a CTA can be found in Secon 6.4. ‘Adding an unauthorised medicinal product in
CTIS’ of this handbook.
When registering medicinal products in the xEVMPD, sponsors are advised to take into account the
publicaon requirements of the relevant CTIS product data fields, as specified in CTIS applicaon fields.
The publicaon modality and melines of product-related fields is defined in Annex I of the Guidance
document on how to approach the protecon of personal data and commercially confidenal
informaon while using the Clinical Trials Informaon System (CTIS). Note that fields taken from xEVMPD
cannot be further amended in CTIS before publicaon. As per the xEVMPD guidance, due to the CTIS
publicaon rules it is recommended that the product name created and entered in xEVMPD does not
include the pharmaceucal form nor the strength of the product.
Training for clinical trial sponsors on how to enter and maintain product informaon into the XEVMPD is
also available (links in the ‘References’ table below).
References
Locaon (area or document)
Data submission on invesgaonal medicines:
guidance for clinical trial sponsors webpage
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-trials/data-
submission-invesgaonal-medicines-guidance-
clinical-trial-sponsors
Guidance on the electronic submission of
informaon on invesgaonal medicinal
products for human use in the Extended
EudraVigilance medicinal product diconary
(XEVMPD)
hps://www.ema.europa.eu/en/documents/other/g
uidance-electronic-submission-informaon-
invesgaonal-medicinal-products-human-use-
extended_en.pdf
Electronic submission of invesgaonal
medicinal product (IMP) data to the Extended
EudraVigilance medicinal product diconary
(XEVMPD) - Frequently asked quesons and
answers (FAQs)
hps://www.ema.europa.eu/en/documents/other/el
ectronic-submission-invesgaonal-medicinal-
product-imp-data-extended-eudravigilance-
medicinal_.pdf
Extended EudraVigilance medicinal product
diconary (XEVMPD) training webpage
hps://www.ema.europa.eu/en/human-
regulatory/post-authorisaon/data-medicines-iso-
idmp-standards/extended-eudravigilance-medicinal-
product-diconary-xevmpd-training
eXtended EudraVigilance Medicinal Product
Diconary (XEVMPD) Data-Entry Tool (EVWEB)
user manual
hps://www.ema.europa.eu/en/documents/other/e
xtended-eudravigilance-medicinal-product-
diconary-xevmpd-data-entry-tool-user-
manual_en.pdf
eXtended EudraVigilance Medicinal Product
Report Message (XEVPRM) Step-by-Step
hps://www.ema.europa.eu/en/documents/other/e
xtended-eudravigilance-medicinal-product-report-
message-step-step-guide-insert-development_en.pdf

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 32/62
Guide: Insert of a Development Medicinal
Product (DMP)
6.2. Medicinal product in CTIS extracted from XEVMPD
For each trial in CTIS, the sponsor has to associate at least one medicinal product with the role as ‘test’
and populate this informaon in the Part I of an inial CTA.
Other product roles that can be associated to a CTA, as applicable, are: comparator, placebo and auxiliary
medicinal product.
In CTIS, the product informaon (for test product, comparator and auxiliary medicinal product) is
retrieved from the XEVMPD and this is enabled by a search and selecon funconality available for an
authorised product (i.e. a product with a markeng authorisaon in the EU/EEA), an acve substance, an
Anatomical Therapeuc Chemicals (ATC) code and an unauthorised product.
6.3. Adding an authorised medicinal product in CTIS
Medicinal product details in an applicaon form are mandatory. The users can add product details in a
CTA for any product role in the trial (test, comparator, auxiliary) by searching and selecng the product
details from XEVMPD. Only for placebo, the product details can be specified locally in CTIS.
A user can add an authorised product by searching per product details, acve substance, or ATC code, as
applicable.
The following parameters are displayed to a user that can search in XEVMPD, via CTIS, for an authorised
medicinal product:

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 33/62
Figure 6.3.1. Search for an authorised medicinal product.
Depending on the parameters used to run the search, the user can get a unique search result returned,
or mulple results can be retrieved.
For example, in case of search by EU MP number (i.e. EV Code assigned to a specific medicinal product
record) only one result will be returned.
However, if the user is searching for an acve substance, a pharmaceucal form or strength of a
medicinal product, then mulple results may be returned.
The following parameters are displayed to a user that can search in XEVMPD, via CTIS, for an acve
substance used in an authorised medicinal product:
Figure 6.3.2. Search for an acve substance.
The following parameters are displayed to a user that can search in XE
VMPD, via CTIS, for an ATC code
(level 3, 4 or 5) associated with an authorised product:
Figure 6.3.3. Search for ATC code (level 3, 4 or 5) associated with an authorised medicinal product.
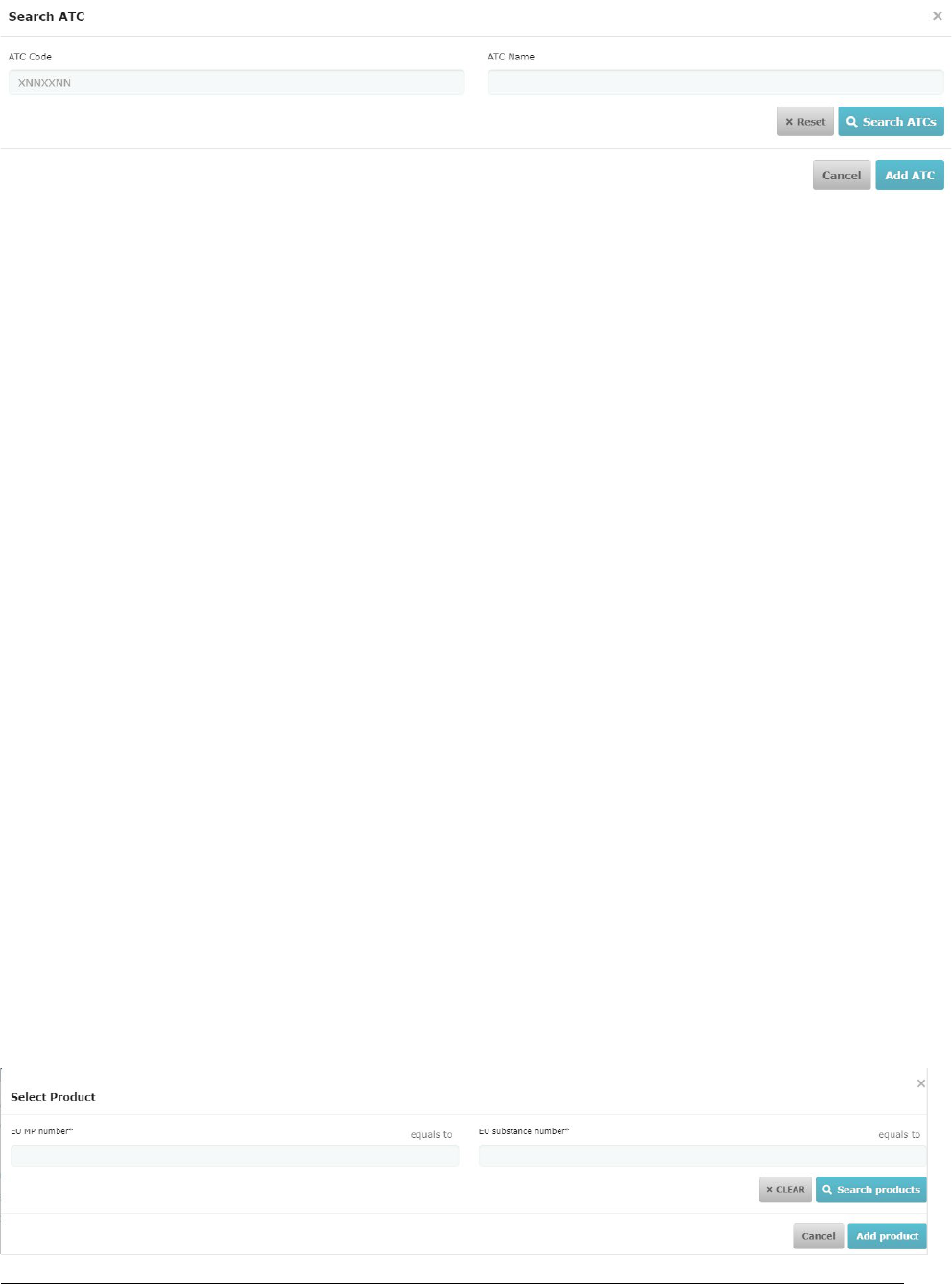
Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 34/62
6.4. Adding an unauthorised medicinal product in CTIS
Unauthorised medicinal product details may contain confidenal informaon and therefore access to
this informaon is restricted.
As explained in Secon 6.1 ‘Medicinal product registraon in XEVMPD’ above, unauthorised products
include those that have not received a markeng authorisaon in the EU/EEA for the strength and/or
pharmaceucal form.
If an acve substance is used in a clinical trial in a new pharmaceucal dose form and/or new strength, a
new development medicinal product must be entered in the XEVMPD by the sponsor organisaon.
If a medicinal product not yet authorised in the EEA is used in a clinical trial for different indicaons
and/or routes of administraon(s), the sponsor can update their exisng development medicinal product
in XEVMPD with the new indicaon/route of administraon.
Registraon of development medicinal products in XEVMPD is independent of the role of the medicinal
product in the clinical trial (i.e. test, comparator etc.) before they can be used to populate dossier part I
of the CTA in CTIS.
Users can retrieve unauthorised products informaon in CTIS only by searching for EU MP number
(medicinal product EV Code) together with the EU substance number (substance EV Code) referenced in
this product in the XEVMPD.
A medicinal product EV Code is a unique number assigned by the XEVMPD to each medicinal product
record successfully inserted in the diconary; it is used to idenfy this medicinal product in the XEVMPD.
It should be noted that both parameters, namely the medicinal product EV Code and the substance EV
Code, are mandatory to run the search in CTIS for unauthorised medicinal product data.
Users have to be cognisant of the required informaon in order to be able to run the search for
development products in XEVMPD and add the product in the CTA of CTIS.
Figure 6.4.1. Search for an unauthorised medicinal product.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 35/62
Once the medicinal product of interest is idenfied, the user will see some pre-populated data, as it is
available in the XEVMPD. The strength and pharmaceucal form of the retrieved unauthorised product
are not displayed in the dra CTA dossier Part I. This informaon only becomes visible following the
submission of the applicaon to the MSCs.
More details on the registraon of medicinal products in XEVMPD are provided in Secon 6.1. of this
handbook.
6.5. Medicinal product details in CTIS
For both authorised and unauthorised products, once the desired medicinal product details are retrieved
from XEVMPD, users will see some pre-populated data in CTIS extracted from XEVMPD.
Some addional details, such as the dosage and administraon details, informaon about the medicinal
product, Advance Therapy Medicinal Product (ATMP) details (as applicable), and combinaon with
medical device (as applicable), will have to be populated in CTIS.
In addion to the populaon of the structured data fields in CTIS, for each product, users also have to
provide the documents foreseen in the Clinical Trials Regulaon, as applicable, namely:
• Invesgator Brochure (IB) or the Summary of Product Characteriscs (SmPC);
• Invesgaonal Medicinal Product Dossier (IMPD) Quality;
• Invesgaonal Medicinal Product Dossier (IMPD) Safety and Efficacy;
• GMP documentaon ;
• Content labelling.
Figure 6.5.1. Addional details for medicinal products (Part I).
For more informaon, see also training module 10.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 36/62
References
Locaon (area or document)
CTIS Training Material Module 10 ‘Create, submit
and withdraw a clinical trial’
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-cs-online-
modular-training-programme#sponsor-
workspace-secon
‘How to submit an inial CTA in the CTIS Sponsor
workspace – Fill in the Part I secon’ (video) -
CTIS Training Material Module 10
hps://www.youtube.com/watch?v=e-JTvFoBlCs
7. Data, documentaon and processes
7.1. Clinical Trial Applicaon (CTA) and Noficaon Forms
This secon intends to provide informaon on the data fields and documents that sponsors need to
complete, as applicable, in the context of clinical trial applicaons and noficaons to be submied to
CTIS. These forms provide an overview of the data fields to be completed, and documents to be provided
with the aim to help sponsors prepare in advance, the informaon required for the submission of an
inial CTA, adding Member State Concerned applicaon (MSC), Substanal Modificaon (SM), non-
Substanal Modificaon, (non-SM) and noficaons.
The forms referred to in this document include the aributes of the fields (e.g. character limit).
7.1.1. Clinical Trial Applicaon Form overview of the data fields to be completed and documents to be
provided
In the table below, there are references to four Excel documents that list the structured data fields and
the documents to be completed. Each data form addresses the informaon to be provided for any type of
clinical trial applicaon, for a mul-trial substanal modificaon, for the response to a request for
informaon, or for an annual safety report. Each document contains an overview with some relevant
instrucons followed by the list of the data fields to be completed, and documents to be uploaded for
each of the CTIS secons to be prepared for an applicaon: the ‘Form’ secon (4 tabs included, one per
applicaon type), ‘MSC’, ‘Part I’ and ‘Part II’ secons. Moreover, the documents include the different
searches that the sponsor will need to perform through interfaces with other systems.
References
Locaon (area or document)
CTIS structured data form - Applicaons (IN,
AMSC, SM, non-SM)
hps://www.ema.europa.eu/documents/template-
form/cs-structured-data-form-inial-applicaon-
addional-member-state-concerned-
substanal_en.xlsx

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 37/62
CTIS structured data form - Applicaons (Mul
trial SM)
hps://www.ema.europa.eu/documents/template-
form/cs-structured-data-form-mul-trial-
substanal-modificaon_en.xlsx
CTIS structured data form – Requests for
Informaon
hps://www.ema.europa.eu/documents/template-
form/clinical-trial-informaon-system-cs-
structured-data-form-request-informaon-
rfi_en.xlsx
CTIS structured data form – Annual Safety
Report
hps://www.ema.europa.eu/documents/template-
form/clinical-trial-informaon-system-cs-
structured-data-form-annual-safety-report-
asr_en.xlsx
7.1.2. Noficaon and Results: overview of the data fields to be completed and documents to be
provided
The document provided in the table below contains an overview with some relevant instrucons
followed by the overview of the data fields to be completed and to be uploaded for each noficaon
form present in the ‘Noficaons’ secon implemented in the system: start of trial, start of recruitment,
end of recruitment, end of trial, global end of trial, temporary halt, restart of trial, restart of recruitment,
ancipated date of summary of results, unexpected event, serious breach, urgent safety measure and 3
rd
country inspectorate inspecon.
Details on the submission of results documents are also provided.
7.1.3. Data fields and document specificaons
Data fields and document specificaons can be found in the overview secon of each CTIS Structured
data form.
When populang clinical trial informaon in CTIS, the following points should be considered:
7.1.3.1. Limits of characters for free-text fields
Generally, there is a limitaon of 4000 characters for manual data free-text fields. Nevertheless, there are
certain fields following masked values, i.e. PIP number (EMEA-111111-PIP11-11) or fields with smaller
sizes. These are further detailed below.
References
Locaon (area or document)
CTIS structured data form – Noficaons and
Results
hps://www.ema.europa.eu/documents/template-form/cs-
structured-data-form-noficaons_en.xlsx

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 38/62
Table 7.1.3.1. Number of characters allowed in free text fields in CTIS
Text field
Character limitaon
Manual data free-text fields (general rule)
4000
Phone number
15
Protocol code, registry idenfier, designaon number for
orphan drug, CAT reference number
20
Sponsor internal idenfier for unexpected event, serious
breach, urgent safety measure, 3
rd
country inspectorate
inspecon noficaons
20
Registry name, source of monetary support, period tle, arm
tle, sponsor contact person first name and last name, address,
town/city, department, email address, product code, gene of
interest, species origin for the xenogeneic cells, ssue-
engineered xenogeneic species of origin, device trade name,
device nofied body, authorisaon number of manufacturing
and import
100
Primary and secondary end points
500
Descripon of the device
2000
7.1.3.2. Characteriscs of documents upload
Every me users upload a document in CTIS, they should keep in mind that the system allows for storage
of clinical trial data with a maximum size of 220 GB and parcularly permits the following characteriscs
for document upload:
Table 7.1.3.2.1. Characteriscs of document upload in CTIS
Document details
System limitaon
Document file name
100 characters. None of these 7 special characters
(/,.;|) allowed
Document version
10 characters, it can be numerical or not
Document comment free text field
4000 characters
Document file size
50 MB
Maximum number of documents uploaded in
one batch
25
References
Locaon (area or document)

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 39/62
Guide on CTIS Common features
hps://www.ema.europa.eu/documents/other/clinic
al-trials-informaon-system-cs-common-features-
cs-training-programme-module-02_en.pdf
CTCG’s Best Pracce Guide for Sponsors of
document naming in CTIS
hps://www.hma.eu/fileadmin/dateien/HMA_joint/
00-_About_HMA/03-
Working_Groups/CTCG/2022_09_CTCG_Instrucon_
naming_documents_CTIS_EU_v1.4.pdf
7.2. Trial categorisaon
The trial category is chosen by the sponsor when filling in the ‘form’ secon of the applicaon, based on
definions provided in table V of Annex I of the
Guidance document on how to approach the protecon
of personal data and commercially confidenal informaon while using the CTIS. Refer to CTIS training
Module 10 e-learning presentaon and FAQs for instrucons on how to change the category of a trial.
References
Locaon (area or document)
CTIS Training Material Module 10 ‘Create,
submit and withdraw a clinical trial’
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-cs-online-
modular-training-programme#sponsor-workspace-
secon
7.3. Download opons
There are two ways for downloading CT and CTAs informaon; either from the clinical trial page or from
the clinical trial applicaon(s) of the trial. There is also an opon for CTIS sponsor users to download
search results when searching for clinical trials. The subsecons below provide users with more details
for each download opon.
7.3.1. Clinical trial page
By clicking on the trial number, users open the clinical trial page and may access all related informaon
submied via the applicaon(s) of the trial. On the upper right corner, users can use the download
buon and retrieve a zip file that includes any informaon related to the trial and its applicaons. The
users, once click on the ’Download’ buon, can view a tree menu from which they can select which data
(applicaon dossier –Form, MSC, Part I and Part II– or Evaluaon related) of an applicaon (only one
applicaon per me can be selected) they wish to include in the downloading zip file. Besides applicaon
related data, users may include in the zip file addional informaon regarding Noficaons, Correcve
Measures and Trial results.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 40/62
Figure 7.3.1.1: Downloading applicaon data from the clinical trial page.
By using the ‘Download’ buon, users can download the latest version of the structured data and
documents that have been submied per applicaon, provided they have permission to access. They can
access and retrieve previous submied versions of data and documents, by navigang through the CTA
pages, as it is described in the following subsecon (second download opon).
7.3.2. Clinical trial applicaon page
Users can download the documents submied in the various secons of a CTA or a non-SM. They can
download the files from the secons Form (e.g. cover leer), Part I (e.g. protocol) and Part II (e.g.
Recruitment arrangements), using the ‘Download’ icon, found on the right side of each document in its
placeholder. Another way to download the documents from the CTA page is to access them from the ‘All
documents’ le, found in the end of the Part I and II secons and use the respecve PDF icons, found on
the right side of all documents.
Figure 7.3.2.1: Downloading documents from the clinical trial applicaon page (‘All documents’ le).

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 41/62
Previous versions of the documents (if any) can be downloaded also. You may access them using the
arrow next to ‘Previous versions’ found on the right side of the documents, and then the ‘Download’
icon.
Figure 7.3.2.2. Downloading previous document versions.
Previous versions of the applicaon and their respecve data can be accessed by using the ‘Versions’
buon, found on the upper right corner of the CTA page.
Figure 7.2.3.2.3: Accessing previous versions of a CTA and respecve data.
7.3.3. Clinical Trial Search Download of results
Within the clinical trial overview page, users can use the subtabs to access and view any data and
informaon related to that clinical trial. One of them, labelled ‘Trial results’, contains the placeholders for
summary of results, for lay person summary of results and for related clinical study reports. The
documents aached to those placeholders can be downloaded by users who have the appropriate
access, by using the respecve download icons. Users may download the data related to clinical trial
results by using the download funconality, found on the CT overview page. By clicking the download
buon, found on the upper right corner, users can select which data to include in their download zip file.
Among the main data groupings, users may retrieve the summary of results/Layperson summary.
References
Locaon (area or document)

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 42/62
Search, view and download informaon on
clinical trials and clinical trial applicaons –
Sponsor users
hps://www.ema.europa.eu/documents/other/step-
step-guide-how-search-view-download-clinical-trial-
clinical-trial-applicaon-authority-cs_en.pdf
7.4. How to apply a change to a clinical trial dossier
7.4.1. During the draing of the inial clinical trial applicaon
Users can edit the applicaon while it is in dra status (i.e. unl it has not been submied). To do so,
users can access the applicaon, from the CT summary page and select the ‘Applicaon ID’ under the
column' ID' of the 'Applicaon and Non-Substanal Modificaon' secon. In order to populate and
upload the relevant informaon and documentaon of an applicaon, users need to click on the padlock
buon of each subsecon. If users need to update documents of an applicaon that is sll in dra, they
need to remove the already aached document and re-upload the new version. They can also edit the
details pertaining to a document. By using the pencil icon, they can make many of the placeholder fields
editable (tle, date, version, comment), and the values populated to them may change. Aerwards, they
can either save the applicaon by clicking on the 'Save' buon in the upper-right corner of the page or, if
all the required fields are completed, submit the applicaon.
References
Locaon (area or document)
‘High-level overview of CTIS workspaces and
common system funconalies’ – CTIS Training
Material Module 02
hps://www.ema.europa.eu/documents/other/clin
ical-trials-informaon-system-cs-common-
features-cs-training-programme-module-
02_en.pdf
7.4.2. Once the inial clinical trial applicaon has been submied
Aer subming the CTA, if users want to update the dossier, they need to create a substanal
modificaon (SM) CTA or a non-substanal modificaon (non-SM), as applicable.
A sponsor can dra and submit a substanal modificaon (SM) or a non-substanal modificaon (non-
SM) to a MSC that does not have an assessment on-going in relaon to this clinical trial (see next table).

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 43/62
Table 7.4.2.1. Allowed submissions whilst ongoing evaluaon of a CTA.
Submission of an
SM to Part I and Part
II
Submission of an SM
to Part I
Submission of an
SM to Part II
Submission of a
non-SM
Submission of an applicaon
for addional MSC
Inial
applicaon
Not unl a decision
is issued by all MSCs
Not unl a decision is
issued by all MSCs
Not unl a
decision is issued
by all MSCs
Not unl a
decision is
issued by all
MSCs
Not unl a decision is issued
by all MSCs
Evaluang Part
I & Part II SM
applicaon
No No No No No
Evaluang Part
I only SM
applicaon
No No No No No
Evaluang Part
II only SM
applicaon
No No Only to MS
who did not
receive the Part
II SM
No yes
Evaluang add
addional
member state
No No
Only to MS not
evaluang the
applicaon to
add an
addional MSC
No Only to MS
that is not an MSC or
evaluang an assessment to
become an MSC
Substanal modificaon (SM) - request by the sponsor for a change of a CT that is likely to substanally
impact the subjects' safety or rights or the reliability/robustness of the generated data. A substanal
modificaon will be evaluated by the MSC aer it has been submied.
The scope of a substanal modificaon can be Part I only, Part II only, or Part I and Part II. Aer selecng
the scope of the SM, users need to prepare the dossier of the SM, adding required informaon, eding
already submied structured data and updang submied documents. They can update a document,
using the ‘update’ icon. Once click on the update icon, users can aach the new version of the document,
indicang their version reference and adding any relevant comment (if needed) in the dedicated free text
field. They can change the informaon pertaining to the document by using the pencil icon and even
empty the document placeholder, by using the remove icon.
Non-substanal modificaon (non-SM) - any change to the CT dossier that is not likely to substanally
impact the safety or rights of the subjects, or the reliability and robustness of the data generated in the
CT but is relevant for the supervision.
Sponsors can submit non-SMs to keep the informaon of the dossier up to date. Non-SM changes can
also be provided as part of RFI responses where so required.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 44/62
A sponsor can only submit an SM or a non-SM to an MSC that does not have an on-going assessment of
an applicaon for the concerned trial. If users need to upload new versions of documents, they can
submit a clean version of the updated document, as well as the original document with tracked changes,
to facilitate the assessment of CTAs.
Table 7.4.2.2. Documents and Structured Data Fields that may be populated with an SM applicaon type.
Form
MSC
Part I
a
Part II
a
Document
for upload
Cover leer
Documents
Documents
Modificaon
descripon
Document
translaons
Document
translaons
Supporng informaon
Proof of payment
Structured
data field
Supporng informaon
Number of
subjects per MSC
Data
Trial site
SM reason
Data translaons
PI contact details
SM scope
Third party enty
Sponsor contact
details
Descripon of
changes
a
If there are any changes to the dossier
Table 7.4.2.2.3. Documents and Structured Data Fields that may be populated with a non-SM applicaon type.
Form
MSC
Part I
a
Part II
a
Document
for upload
Documents
b
Documents
Document
translaons
Document
translaons
Structured
data field
Modificaon
descripon
Number of subjects
per MSC
Data
b
PI contact details
Data translaons
Third party enty

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 45/62
Form
MSC
Part I
a
Part II
a
Sponsor contact
details
Descripon of
changes
a
If there are any changes to the dossier
b
Document/Field can be modified with limitaons
References
Locaon (area or document)
How to Manage a CT – CTIS Training Material
Module 05 eLearning
Secon 3 - Update and withdraw other types of
noficaons slide
hps://www.ema.europa.eu/en/learning-module/manage-
ct/story.html
hps://www.ema.europa.eu/en/documents/other/s
tep-step-guide-how-search-view-download-clinical-
trial-clinical-trial-applicaon-sponsors-cs_en.pdf
Create, submit and withdraw a CTA – CTIS
Training Material Module 10 eLearning
Secon 2 – Edit and upload
hps://www.ema.europa.eu/en/learning-
module/create-ct-applicaon/story.html
‘Step-by-step guide’ – CTIS Training Material
Module 10
Page 4
hps://www.ema.europa.eu/en/documents/other/s
tep-step-guide-create-submit-withdraw-clinical-trial-
applicaon-nonsubstanal-modificaons-cs_en.pdf
FAQs - CTIS Training Material Module 10
Quesons
1.6. How can users edit a CTA?
2.3. How can users create & edit an Inial CTA?
hps://www.ema.europa.eu/en/documents/other/f
aqs-how-create-submit-withdraw-clinical-trial-
applicaon-cs-training-programme-module-
10_en.pdf
7.5. Handling of Requests for Informaon (RFIs) in CTIS
During the evaluaon of CTAs, MSC have the possibility to require clarificaons from the sponsors by
raising RFIs that should be addressed within the defined melines. It should be noted that failing to
provide responses within the melines will lead to the applicaon being lapsed. It is encouraged that
high-quality dossiers are submied in CTIS with each applicaon, to minimise, where possible, the need
to raise a request for informaon.
RFI can be idenfied by the sponsors via monitoring the noces and alerts tab and the RFI tab in CTIS
Sponsor workspace. For example, in an inial applicaon with Part I and Part II, an RFI can be raised by
the RMS as part of the validaon and assessment of Part I and by each MSC following art II assessment.
RFI can be raised by the RMS and MSC at any point in me during the evaluaon phase. There are no
predicted melines and period of me when RFI can be raised, therefore the sponsors should be vigilant
in monitoring the noces and alerts and the RFI tab.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 46/62
RFIs are raised by the RMS/MSC via the consideraons documented in the system as part of the
evaluaon. Documented consideraons are then consolidated by the RMS/MSC
(accepted/merged/adapted or rejected) directly in CTIS and used as the basis for the RFI. RMS/MSC can
also upload documents into CTIS as supporng documentaon to the RFI being raised.
Sponsors have the possibility to download from CTIS the consideraons part of the RFI, as well as any
supporng documentaon, so the RFI can be allocated to relevant team members to be addressed. Users
can also have access to the consideraons in the RFI and any documents directly from CTIS and provide
their reply directly in the system.
In order to address an RFI, the sponsor has to provide a response in the free text displayed aer each
consideraon raised by the RMS/MSC as part of the RFI, that can be complemented by supporng
documents. RFI responses can be saved as dras before submission.
Figure 7.5.1. Response to an RFI consideraon (free text with oponal supporng documents) can be saved as dra before submission.
The sponsor also has the possibility to apply changes to the dossier, both structure data and documents,
depending on the nature of the request raised. If changes to the dossier are applied, the sponsor should
also provide a document containing a descripon of the changes made.
Figure 7.5.2. Changes to a CTA dossier shall be described in a document.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 47/62
Previously uploaded documents can be deleted when responding to an RFI or a new version of a
document can be provided (in this case documents will have the same document type, language and
tle). When uploading a new document, the sponsor can specify the date and the version of the file, and
a system version will also be generated sequenally by the system, independently of the sponsor’s
version number.
Figure 7.5.3. An RFI response may include uploading, updang or deleng a document.
Completely new documents can also be submied when replying to an RFI, for the secon of the
applicaon dossier in queson and subject to the RFI.
Access to the RFI, as well as download funconality, depends on the user profile. For example, in Part I
users will only have access to RFI pertaining to Part I of the dossier. Each user can download the RFI and
RFI responses that the user has access to. It should be noted that CT Administrators can assign to
themselves access to Part I and Part II and therefore can have access to all RFIs.
CTIS enables sponsors users to address RFIs simultaneously in the different secons of the CTA, namely a
user can work on Part I RFI at the same me as users working on Part II RFI. Also, Part II RFIs raised by
different MSCs can be addressed simultaneously by different users, if needed. In case of simultaneous
RFIs, users should be mindful to work on their respecve RFIs.
Figure 7.5.4. New CTA dras are created to respond to RFIs simultaneously.
RFI raised in the course of evaluang a Clinical Trial Applicaon (CTA) and the responses provided are
subject to publicaon rules, except for RFI raised for secons of the applicaon that are exempted from
publicaon, such as the quality secon of the dossier or quesons related to quality in general.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 48/62
Training material on how to address incoming RFIs related to the evaluaon of a CTA has been published
on the CTIS Training Material Catalogue, Module 11.
As part of an ad hoc assessment, in case of evaluaon of an annual safety report (ASRs), or when a
sponsor opinion needs to be provided in the context of a correcve measure, an RFI can also be raised.
That RFI and its response are exempt from publicaon.
Training material on how to address other types of incoming RFIs (Ad hoc assessment, Correcve
measures) has been published in the CTIS Training Material Catalogue, Modules 04 and 05. Training
material on how to address incoming RFIs related to ASR is published in the CTIS Training Material
Catalogue, Module 18.
References
Locaon (area or document)
CTIS Training Material Module 04 ‘Support
with workload management in the sponsor
workspace: eLearning – RFI secon’
hps://www.ema.europa.eu/en/learning-
module/workload-management-sponsor/story.html
CTIS Training Material Module 04 ‘Support
with workload management in the sponsor
workspace: FAQs – RFI secon’
hps://www.ema.europa.eu/en/documents/other/faqs-
support-workload-management-workspace-cs-
training-programme-module-04_en.pdf
CTIS Training Material Module 04 ‘Support
with workload management in the sponsor
workspace: Videoclip’
hps://www.youtube.com/watch?v=6z-Q6-LK8Ws&ab
CTIS Training Material Module 05 ‘Manage
a CT through CTIS: eLearning – various
references to types of RFI’
hps://www.ema.europa.eu/en/learning-
module/manage-ct/story.html
CTIS Training Material Module 05 ‘Manage
a CT through CTIS: FAQs – various
references to types of RFI’
hps://www.ema.europa.eu/en/documents/other/faqs-
how-manage-ct-cs-training-programme-module-
05_en.pdf
CTIS Training Material Module 11 ‘Respond
to requests for informaon received during
the evaluaon of a clinical trial applicaon:
eLearning’
hps://www.ema.europa.eu/en/learning-
module/respond-to-rfis-cs/story.html
CTIS Training Material Module 11 ‘Respond
to requests for informaon received during
the evaluaon of a clinical trial applicaon:
FAQs’
hps://www.ema.europa.eu/en/documents/other/faqs-
how-respond-requests-informaon-received-during-
evaluaon-clinical-trial-applicaon-cs_en.pdf
CTIS Training Material Module 11 ‘Respond
to requests for informaon received during
the evaluaon of a clinical trial applicaon:
Videoclips’
hps://www.youtube.com/watch?v=vbQVkYi3pGI&ab
hps://www.youtube.com/watch?v=DXrQMStp2a0&ab
hps://www.youtube.com/watch?v=sO8YRSatsDA&ab
CTIS Training Material Module 18 ‘How to
create and submit an annual safety report
hps://www.ema.europa.eu/en/documents/other/step-
step-guide-how-create-submit-annual-safety-report-
respond-related-requests-informaon-cs_en.pdf

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 49/62
and respond to related requests for
informaon: Step by Step guide’
7.6. Subming the Clinical Study Report (CSR)
Refer to the following Quick guide for informaon on how to submit a CSR.
References
Locaon (area or document)
CTIS Training Material Module 13 ‘Clinical
Study Reports submission Quick guide’
hps://www.ema.europa.eu/en/documents/other/quick-
guide-clinical-study-reports-submission-cs-training-
programme-module-13_en.pdf
8. Data transparency
The clinical trial informaon processes and flows in CTIS start with a CTA submied by the sponsor, or
delegated enes, via CTIS secure domain (see Secon 4. ‘How to get a clinical trial applicaon started in
CTIS ’), to carry out a clinical trial in the EU/EEA, and the corresponding evaluaon performed by the
MSCs.
Following this evaluaon, a decision is issued by each MSC for the CTA, on whether the trial is authorised,
authorised with condions, or not authorised. Aer a decision of any kind has been issued by the MSC,
data and documents submied to CTIS for the trial will be made available to the public as per the
modality and melines defined in the Annex 1 to the
Guidance document on how to approach the
protecon of personal data and commercially confidenal informaon while using the CTIS. The detailed
list of structured data that are, or are not subject to publicaon is specified in the files menoned in
Secon
7.1. of this document (specifically: CTIS applicaon fields and Noficaons and Results). A useful
summary of the rules can be found in the quick user guide. All the menoned reference documents
reflect the current publicaon rules for CTIS, defined in the Revised CTIS transparency rules, and provide
guidance on the protecon of personal data and commercially confidenal informaon (CCI) submied
to the system, in accordance with the requirements of Arcle 81(4) of Regulaon (EU) No 536/2014
(CTR). A Quesons and Answers (Q&A) document on this topic is also available to users, see Q&A o
n the
protecon of Commercially Confidenal Informaon and Personal Data while using CTIS.
The disclosure melines of data in CTIS depend on the trial category, on the populaon age (in case of
category 1 trials) and on the trial phase (in case of category 2 trials that are integrated phase 1 and 2).
The trial category is chosen by the sponsor when filling in the ‘form’ secon of the applicaon, based on
definions provided in Table V of Annex I
. Excepons to these disclosure rules apply to all trials submied
before 18 June 2024 (referred to as ‘historical trials’), which have only their structured data published;
moreover, for those trials documents submied through part I Non-Substanal Modificaons and
addional member state applicaons are also not published: see secon 2.3 of
Guidance document and
table IV of its Annex I.
For all documents that are in scope of publicaon (see Table II of Annex I of the guidance document),
users need to provide a document version ‘for publicaon’, where personal data and CCI should be
properly redacted or should not appear (see chapter 3 and 4 of the
Guidance document). Please note

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 50/62
that any document inadvertently uploaded ‘for publicaon’ into the relevant CTIS document upload
secons will be published. For example, if an IB is uploaded into the SmPC secon of a Category 2 or 3
trial, the IB will be made public if the sponsor does not correct this oversight before the decision on the
applicaon.
The ability to upload a version ‘not for publicaon’ is made available to the sponsor, in order to provide
informaon on personal data and CCI that are deemed necessary for the assessment by the RMS/MSCs.
It is not expected that such a version is provided by the sponsor for all document types: this need
depends on the document content. This funconality allows the exchange of informaon in the CTIS
secure domain between users with regulated access depending on their profile, while at the same me
protecng personal data and the legimate interest of sponsors for what concerns CCI.
With respect to those documents that are not subject to publicaon, note that quality and non-quality
documents need to be submied as separate documents. This includes the IMPD-Q, Scienfic Advice -
Quality and Quality RFI response documents. CTIS user roles depend on maintaining separate quality
documents to ensure only authorised users can view quality informaon.
References
Locaon (area or document)
Regulaon (EU) No 536/2014 of the European
Parliament and of the Council of 16 April 2014 on
clinical trials on medicinal products for human
use, and repealing Direcve 2001/20/EC
hps://eur-lex.europa.eu/legal-
content/EN/TXT/?uri=celex%3A32014R0536
Revised CTIS transparency rules
hps://www.ema.europa.eu/en/documents/other
/revised-cs-transparency-rules_en.pdf
Guidance document on how to approach the
protecon of personal data and commercially
confidenal informaon while using the Clinical
Trials Informaon System (CTIS)
hps://accelerang-clinical-
trials.europa.eu/document/download/6a0b836f-
4779-4bb9-9584-
1ce504a9ae38_en?filename=guidance-document-
how-approach-protecon-personal-data-
commercially-confidenal-informaon-while_.pdf
Annex I to the Guidance document
hps://accelerang-clinical-
trials.europa.eu/document/download/824905dd-
3033-41e6-a871-
67b20c4f4c94_en?filename=annex-i-guidance-
document-how-approach-protecon-personal-
data-commercially-confidenal_.pdf
Q&A on the protecon of Commercially
Confidenal Informaon and Personal Data while
using CTIS
hps://accelerang-clinical-
trials.europa.eu/document/download/33702a5d-
13be-4c4f-936d-
3627dd73085b_en?filename=ACT%20EU_Q%26A%
20on%20protecon%20of%20Commercially%20Co
nfidenal%20Informaon%20and%20Personal%20
Data%20while%20using%20CTIS_v1.3.pdf

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 51/62
Quick user guide
hps://accelerang-clinical-
trials.europa.eu/document/download/a101771b-
0be7-492f-b8bd-
7f551b7a7_en?filename=Revised%20CTIS%20tr
ansparency%20rules%2C%20Interim%20period%20
%26%20Historical%20trials_quick%20guide%20for
%20users_1.pdf
CTIS structured data form - Applicaons (IN,
AMSC, SM, non-SM)
hps://www.ema.europa.eu/documents/template-
form/cs-structured-data-form-inial-applicaon-
addional-member-state-concerned-
substanal_en.xlsx
CTIS structured data form – Noficaons and
Results
hps://www.ema.europa.eu/documents/template-
form/cs-structured-data-form-
noficaons_en.xlsx
CTIS Training Material Module 12 ‘Data
protecon: e-learning course’
hps://www.ema.europa.eu/en/learning-
module/data-protecon-cs/story.html
9. Safety reporng obligaons
9.1. Suspected Unexpected Serious Adverse Reacons (SUSARs)
The reporng of SUSARs by the sponsor to the European Medicines Agency in the context of the CTR is
outlined in Arcle 42. The most relevant change for sponsors is the legal obligaon for the electronic
reporng of SUSARs to the clinical trial module of EudraVigilance for a CT performed in at least one
Member State (Art 42.1).
Where a sponsor, due to a lack of resources, does not have the possibility to report to EudraVigilance,
and the sponsor has the agreement of the MSC, it may report to the Member State where the SUSARs
occurred. That Member State shall then report the SUSARs to EudraVigilance (Art 42.3).
Since CTIS was launched, CT-3 final arrangement for SUSARs applies also to all trials approved
through the CTD, as announced by the Clinical Trials Expert Group (CTEG) in April 2021. This means
that from 31 January 2022, sponsors report SUSARs to EudraVigilance only and it is no longer
required to send them to member states, regardless of whether the trial has been approved
through the CTR or CTD. This brings the benefit of a single submission process and harmonised
procedures to the area of SUSAR reporting. Member states have the ability to set up SUSAR rerouting
rules in EudraVigilance if they wish to receive copies of SUSARs for their national systems. This applies
for all trials approved under the CTD or CTR.
For some member states, continued direct SUSAR reporting to ethics committees for clinical trials
under the CTD is still also required under their national law; thus, this may be considered on top of the
direct reporting to EudraVigilance. In case of doubt, liaise directly with the member state concerned
(for a link to the member states contact points, refer to Section 11 ‘Other references’).
Updated informaon regarding reporng safety informaon on clinical trials can be found on the EMA
webpage.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 52/62
Secon 7c (REPORTING OF ADVERSE EVENTS/REACTIONS) of the EudraLex Volume 10 Q&A published by
the Commission addresses some addional quesons in the context of SUSAR reporng.
References
Locaon (area or document)
Regulaon (EU) No 536/2014 of the European
Parliament and of the Council of 16 April 2014 on
clinical trials on medicinal products for human
use, and repealing Direcve 2001/20/EC
hps://eur-lex.europa.eu/legal-
content/EN/TXT/?uri=celex%3A32014R0536
Link to EudraLex - Volume 10 - Clinical trials
guidelines - Set of documents applicable to
clinical trials authorised under Regulaon EU No
536/2014
8
hps://health.ec.europa.eu/medicinal-
products/eudralex/eudralex-volume-10_en#set-of-
documents-applicable-to-clinical-trials-authorised-
under-regulaon-eu-no-5362014
Detailed guidance on the collecon, verificaon
and presentaon of adverse events/reacons
arising from clinical trials on medicinal products
for human use (‘CT-3’)
hps://eur-lex.europa.eu/legal-
content/EN/TXT/?uri=CELEX:52011XC0611(01)
CTEG announcement that CT-3 final
arrangements for SUSAR reporng will apply
since CTIS launch
hps://ec.europa.eu/transparency/expert-groups-
register/core/api/front/document/56534/download
Reporng safety informaon on clinical trials
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/reporng-safety-informaon-clinical-trials
EudraVigilance: electronic reporng
hps://www.ema.europa.eu/en/human-
regulatory/research-
development/pharmacovigilance/eudravigilance/eudrav
igilance-electronic-reporng
9.2. Annual Safety Report (ASR)
The reporng of ASRs by the sponsor to the Agency in the context of the CTR is outlined in Arcle 43,
applicable to trials registered in CTIS and managed under the CTR. The sponsor shall submit annually
through CTIS a report on the safety of each invesgaonal medicinal product (IMP) used in a clinical trial,
other than placebo, for which it is the sponsor. This obligaon referred to in paragraph 1 starts with the
first authorisaon of a clinical trial in accordance with the CTR and it ends with the end of the last clinical
trial conducted by the sponsor with the IMP.
Secon 7d (ANNUAL SAFETY REPORTS) of the Q&A published by the Commission in EudraLex Volume 10
(see ‘References’ table above) may address some of the quesons in the context of ASR reporng.
8
The latest published European Commission Clinical Trials Regulaon No 536/2014 Q&A document can be found under the ‘Set of documents applicable
to clinical trials authorised under Regulaon EU No 536/2014’ secon.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 53/62
The documents referred to in this handbook’s Secon 9.2. apply also to this secon. In addion, you can
find the link to the ASR submission training module below:
References
Locaon (area or document)
CTIS Training Material Module 18 ‘How to
create and submit an annual safety report and
respond to related requests for informaon’
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-cs-online-
modular-training-programme#sponsor-workspace-
secon
CTIS Training Material Module 18 ‘How to
create and submit an annual safety report and
respond to related requests for informaon:
Step by Step Guide’
hps://www.ema.europa.eu/en/documents/other/st
ep-step-guide-how-create-submit-annual-safety-
report-respond-related-requests-informaon-
cs_en.pdf
10. Support
10.1. Release notes and known issues
EMA regularly performs technical updates to CTIS to improve its features and funconality. When
significant updates are made to CTIS, EMA publishes release notes that outline what has changed in the
system. Updates may include improvements to exisng features and funconality, the addion of new
features as well as funconality and technical improvements.
In addion, EMA publishes known issues that sponsor and authority users may encounter when using the
CTIS secure workspaces. Where possible, workarounds to apply are proposed.
All versions of the release notes and known issues documents can be found on the ‘Website outages and
system releases’ page of EU Clinical Trials. CTIS users are advised to make use of the latest version of the
lists of known issues published on this page.
References
Locaon (area or document)
EU Clinical Trials Website – Website outages and
system releases
hps://euclinicaltrials.eu/website-outages-and-
system-releases
10.2. CTIS Highlights Newsleers
To stay up to date with developments and plans, see EMA Clinical Trials Highlights Newsleers on EMA
corporate website: subscribe at hps://ec.europa.eu/newsroom/ema/user-subscripons/3201/create
References
Locaon (area or document)

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 54/62
Clinical Trials Highlights Newsleer
hps://www.ema.europa.eu/en/news-
events/publicaons/newsleers#clinical-trials-
highlights-secon
10.3. CTIS informaon events
The events page on the EMA corporate website displays informaon events organised by EMA on CTIS
(search words e.g. CTIS; SME).
In order to further support CTIS sponsor users aer CTIS go-live, EMA has launched series of virtual
events where sponsors can ask their quesons live to CTIS experts. CTIS walk-in clinics
provide an
opportunity for sponsors to receive praccal guidance about the Clinical Trials Informaon System by
asking quesons to CTIS experts in real-me.
CTIS bitesize talks are themed events offering users live
system demonstraons on a specific CTIS funconality on each session, and quesons are also received
and answered by CTIS experts in real-me. In addion, EMA introduced OMS troubleshoong sessions
for CTIS users, another series of virtual events aiming to address and clarify outstanding issues and
quesons related to registering organisaon and/or locaon data in OMS for use in CTIS CTAs.
All of these events are open to everyone. The recorded videos of past events become available for the
public on EMA’s YouTube channel and can be accessed also through the dedicated event pages.
For a complete list of the CTIS-related virtual events, sessions and webinars, visit the EMA’s webpage on
CTIS Training and informaon events.
References
Locaon (area or document)
EMA corporate website Events page
hps://www.ema.europa.eu/en/events/upcoming-
events
EMA CTIS Training and informaon events page
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-training-
support#training-and-informaon-events-secon
10.4. CTIS training
Training from EMA on how to use the Clinical Trials Informaon System (CTIS) is available. The EMA’s
training resources are tailored for clinical trial sponsors and staff of the European Union (EU) Member
States, European Commission and other organisaons who will use the system.
The EMA CTIS training programme is mainly composed of online training modules published on the CTIS
training programme page on the EMA corporate website. A wide selecon of materials in different
formats are available on introductory modules, common funconalies for all registered users, modules
on the authority (Member States, EMA and European Commission) workspace and on the sponsor
workspace. It also includes recordings from virtual training sessions organised by EMA as well as a secon
with informaon about the Master trainer programme.
When starng to use the training materials, it is advised that organisaons and users first make use of the
Guide to CTIS Training Material Catalogue.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 55/62
Reproducon and/or distribuon of the content of the published training materials is authorised for non-
commercial or commercial purposes, provided that the EMA is acknowledged as the source of the
materials.
EMA has developed the training materials to enhance public access to informaon on CTIS. The training
materials describe inially a preliminary version of CTIS and while the material will undergo revision it
may therefore, not enrely describe the system as it is at the me of use of the material. The Agency
does not warrant or accept any liability in relaon to the use (in part or in whole) or the interpretaon of
the informaon contained in this training material by third pares.
Limited end user training events are organised by EMA and announcements are made on the events page
(see ‘References’ table in 10.4) of the EMA corporate website (search e.g. with word ‘CTIS’ or ‘SME’).
References
Locaon (area or document)
Clinical Trials Informaon System (CTIS) online
training modules page (EMA corporate website)
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/training-support/clinical-trials-informaon-
system-cs-online-training-modules
Guide to CTIS Training Material Catalogue (EMA
corporate website)
hps://www.ema.europa.eu/en/documents/other
/guide-cs-training-material-catalogue_en.pdf
10.5. CTIS training environment for user training and organisaon preparedness
The Clinical Trial Information System training environment (CTIS Sandbox) is a copy of a recent
version of CTIS albeit not always identical to the latest version. The purpose of CTIS Sandbox is to
enable knowledge acquisition of the already implemented functionalities of CTIS, by the future CTIS
users and their organisations in a practical way and in a safe environment. CTIS Sandbox use is
directed by conditions, instructions and guidance, and is made available by EMA. EMA will maintain
support for the CTIS Sandbox users through a CTIS Training Environment Support Service (TESS).
Access to CTIS Sandbox is based on need and is therefore intended for, and limited to, those
individuals and organisations that are the already using or intending to use the secure workspaces of
CTIS (authority and sponsor workspace).
A phased rollout of the CTIS Training Environment is provided to defined user groups in sequence,
started with Member States and the European Commission and followed by Sponsors.
Currently, access to the CTIS Training Environment is provided to representatives of sponsor
organisations based on the need to create an initial clinical trial application. Organisations have been
offered the opportunity to access the CTIS Training Environment in several phases before and after
CTIS go-live.
Access to the CTIS Training Environment can be requested by completing a self-assessment through a
survey (Survey) that collects information on contact details of individuals, the organisations that they
represent, and their plans/need for the use of CTIS. The data collected allows EMA to understand the
need for access to CTIS Training Environment and to consider when access will be granted to ensure
that the Agency is able to support the CTIS Training Environment users effectively. Organisations
wishing to express interest for access to the CTIS Training Environment after the closure of the Survey,

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 56/62
can stay tuned for latest updates on the Survey by subscribing to the Clinical Trials Highlights
Newsletters (see Section 10.2).
Before accessing the CTIS Training Environment, users need to be thoroughly trained on CTIS to get
the best out of their access. The EMA’s published online modular training program can be used for such
preparation purposes.
Version deployments and planned downtimes of the CTIS Training Environment are communicated to
users as required.
References
Locaon (area or document)
CTIS Training Environment Survey
hps://ec.europa.eu/eusurvey/runner/2abb5ba8-
0ec4-9979-b692-0c63f4508b9b
CTIS Training Environment Support Service (TESS)
hps://support.ema.europa.eu/
10.6. Quesons and answers on CTR, CTIS and other EMA IT systems
Quesons and answers on the Clinical Trials Regulaon are available in EudraLex Volume 10 Q&A.
Frequently Asked Quesons on CTIS funconalies are available as part of the published online training
material modules.
If answers cannot be found, sponsors with an EMA account can direct their queries to the EMA’s Service
Desk on CTIS. For sponsors that have not yet created an EMA account, general quesons on CTIS
funconalies can be directed through AskEMA by use of the general form.
For technical support with other EMA IT systems (e.g. EudraVigilance, IRIS, EudraCT), use the EMA
ServiceNow portal (see table below for links).
References
Locaon (area or document)
Quesons & Answers on CTR - EudraLex
Volume 10 - Clinical trials guidelines
9
hps://health.ec.europa.eu/medicinal-
products/eudralex/eudralex-volume-10_en#set-of-
documents-applicable-to-clinical-trials-authorised-under-
regulaon-eu-no-5362014
Frequently Asked Quesons (FAQs) on CTIS
funconalies
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-trials/clinical-
trials-informaon-system-cs-online-modular-training-
programme
FAQs are available within each respecve training module
9
The latest published European Commission Clinical Trials Regulaon No 536/2014 Q&A document can be found under the ‘Set of documents applicable
to clinical trials authorised under Regulaon EU No 536/2014’ secon.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 57/62
EMA Service Desk for CTIS: Quesons on
CTIS funconalies - for EMA account
holders
hps://support.ema.europa.eu/esc?id=emp_taxonomy_t
opic&topic_id=2111dcb6c39d9d10e68bf1f4e40131ee
AskEMA: Quesons on CTIS funconalies
- for non-EMA account holders
hps://www.ema.europa.eu/en/about-us/contact/send-
queson-european-medicines-agency
EMA ServiceNow: Assistance with
informaon technology (IT) systems
hps://support.ema.europa.eu/esc
Q&A on the protecon of Commercially
Confidenal Informaon and Personal Data
while using CTIS
hps://accelerang-clinical-
trials.europa.eu/document/download/33702a5d-13be-
4c4f-936d-
3627dd73085b_en?filename=ACT%20EU_Q%26A%20on
%20protecon%20of%20Commercially%20Confidenal%
20Informaon%20and%20Personal%20Data%20while%2
0using%20CTIS_v1.3.pdf
Quesons and answers – Clinical Trials
Informaon System (CTIS) and Clinical Trials
Regulaon (CTR)
hps://www.ema.europa.eu/en/documents/other/ques
ons-answers-query-management-working-group-cs-
ctr_en.pdf
CTCG Q&A on submission Complex Clinical
Trials in CTIS, vs 1.0, dd 14 March 2023
hps://www.hma.eu/fileadmin/dateien/HMA_joint/00-
_About_HMA/03-
Working_Groups/CTCG/2023_03_CTCG_QA_complex_cli
nical_trials_and_CTIS_v1.0.pdf
Complex clinical trials – Quesons and
answers
hps://health.ec.europa.eu/system/files/2022-
06/medicinal_qa_complex_clinical-trials_en.pdf
10.7. Support for SME and academia sponsors
Specific events and dedicated training materials are organised for SME and academia sponsors. For more
informaon on future events and past recordings, users can visit ‘CTIS Training and informaon events’
webpage.
References
Locaon (area or document)
CTIS Training Module 19 ‘CTIS for SMEs and
academia’
hps://www.ema.europa.eu/en/documents/other/q
uick-guide-introducon-cs-smes-academia-cs-
training-programme-module-19_en.pdf
CTIS Training and informaon events
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-training-
support#training-and-informaon-events-secon

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 58/62

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 59/62
11. Other references
References
Locaon (area or document)
Clinical Trials Informaon System – EMA
webpages
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system
hps://www.ema.europa.eu/en/human-
regulatory/research-development/clinical-
trials/clinical-trials-informaon-system-training-
support
Clinical Trials in the European Union
hps://euclinicaltrials.eu/home
Regulaon (EU) No 536/2014 of the European
Parliament and of the Council of 16 April 2014 on
clinical trials on medicinal products for human
use
hps://eur-lex.europa.eu/legal-
content/EN/TXT/?uri=celex%3A32014R0536
Link to Commission website containing
informaon on clinical trials in the context of
Regulaon EU No 536/2014
hps://ec.europa.eu/health/human-use/clinical-
trials/regulaon_en
Link to EudraLex - Volume 10 - Clinical trials
guidelines
hps://ec.europa.eu/health/documents/eudralex/vo
l-10_en#fragment1
List of naonal contact points
(frequently updated)
Last document in Chapter V of Eudralex vol.10 CTR
(see above)
EudraCT (European Union Drug Regulang
Authories Clinical Trials Database)
10
hps://eudract.ema.europa.eu/
CTIS website outages and system releases
hps://.eu/website-outages-and-system-releases
Conclusion of VHP Procedure - Deadline for
submissions to VHP in the context of the
Christmas Break 2021/2022 and transion to
CTIS/CTR starng with the CTR applicaon
hps://www.hma.eu/fileadmin/dateien/Human_Me
dicines/01-
About_HMA/Working_Groups/CTFG/2021_07_CTFG
_Conclusion_VHP_Deadlines_for_VHP_Submissions.
pdf
ACT EU website: Implementaon of the Clinical
Trials Regulaon
hps://accelerang-clinical-trials.europa.eu/priority-
acon-areas/implementaon-clinical-trials-
regulaon_en
10
EudraCT is the European database for intervenonal clinical trials on medicinal products authorized in the European Union (EEA) and outside the
EU/EEA if they are part of a Paediatric Invesgaon Plan (PIP) from 1 May 2004 onwards; established in accordance with Direcve 2001/20/EC.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 60/62
12. Acronyms and Glossaries
References
Locaon (area or document)
CTIS Training: List of Acronyms
hps://www.ema.europa.eu/en/documents/other/c
s-training-list-acronyms_en.pdf
EMA General Glossary of regulatory terms
hps://www.ema.europa.eu/en/about-us/about-
website/glossary
EMA Medical Terms Simplifier
hps://www.ema.europa.eu/en/documents/other/e
ma-medical-terms-simplifier_en.pdf
Acronym
Term
Definion
CAT
Commiee for Advanced
Therapies
The commiee that is responsible for assessing the quality,
safety and efficacy of advanced therapy medicines,
including medicines classified as gene therapy, somac cell
therapy or ssue-engineered products.
CCI
Commercially confidenal
informaon
Informaon whose publicaon might prejudice the
commercial interests of individuals or companies to an
unreasonable degree. The Agency cannot disclose
commercially confidenal informaon unless there is an
overriding public interest in disclosure.
CTD
Clinical Trial Direcve
2001/20/EC
Introduced to simplify and harmonise the administrave
provisions governing clinical trials in Europe. It was
repealed by the Clinical Trial Regulaon applicaon.
CTEG
Clinical Trials Experts Group
CTFG &
CTFG
Clinical Trials Facilitaon
Group
Acts as a forum for discussion to agree on common
principles and processes to be applied throughout the
European medicines regulatory network (EMRN). It also
promotes harmonisaon of clinical trial assessment
decisions and administrave processes across the naonal
competent authories (NCAs).
CTR
Clinical Trial Regulaon
European Union (EU) pharmaceucal legislaon known as
the Clinical Trials Regulaon entered into applicaon on 31
January 2022. It aims to ensure the EU offers an aracve
and favourable environment for carrying out clinical
research on a large scale, with high standards of public
transparency and safety for clinical trial parcipants.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 61/62
DMP
Development Medicinal
Product
A medicinal product under invesgaon in a clinical trial in
the EEA which does not have a markeng authorisaon in
the EEA and to which special confidenality arrangements
need to be applied.
DSMB
Clinical Trial Data Safety
Monitoring Board
A group of independent individuals, external to the trial,
who are experts in relevant areas. They review the
accumulated data from one or more ongoing clinical trials
on a regular basis and advise the sponsor about the
connued safety of the trial parcipants, the connued
validity of the trial, and the connued scienfic merit of
the trial.
EMA
European Medicines
Agency
Agency of the European Union in charge of the evaluaon
and supervision of pharmaceucal products.
EU
European Union
Supranaonal polical and economic union of 27 member
states that are located primarily in Europe.
EU MP
number
EU Medicinal Product
number
Idenfier issued by the European Medicines Agency for
treatments approved in the European Union.
EUDRACT
European Union Drug
Regulaon Authories
Clinical Trials Database
EudraCT (European Union Drug Regulang Authories
Clinical Trials) is the European Clinical Trials Database of all
intervenonal clinical trials of medicinal products
commencing in the European Union from 1 May 2004
onwards. The EudraCT database has been established in
accordance with Direcve 2001/20/EC.
EV code
EudraVigilance code
EudraVigilance Code
EVWEB
EudraVigilance web-based
tool
EVWEB allows the sending and receiving of safety and
acknowledgement messages in compliance with the latest
ICH M2 standards.
FAQ
Frequently Asked Quesons
A queson in a list of quesons and answers intended to
help people understand a parcular subject.
IB
Invesgator Brochure
A mulfunconal regulatory document essenal for the
conduct of clinical trials that summarises the physical,
chemical, pharmaceucal, pharmacological, and
toxicological characteriscs of an invesgaonal medicinal
product (IMP) as well as any clinical experience.
MAA
Markeng Authorisaon
Applicaon
An applicaon made to a European regulatory authority
for approval to market a medicine within the European
Union.

Clinical Trials Informaon System (CTIS) - Sponsor Handbook
EMA/923413/2022
Page 62/62
PI
Principal Invesgator
The person(s) in charge of a clinical trial or a scienfic
research grant. The PI prepares and carries out the clinical
trial protocol (plan for the study) or research paid for by
the grant. The PI also analyses the data and reports the
results of the trial or grant research.
PIP
Paediatric invesgaon plan
A development plan aimed at ensuring that the necessary
data are obtained to support the authorisaon of a
medicine for children, through studies in children. All
applicaons for markeng authorisaon for new medicines
have to include the results of studies as described in an
agreed paediatric invesgaon plan, unless the medicine is
exempt because of a deferral or waiver.
SME
Micro, Small to Medium-
size Enterprise
SmPC
Summary of Product
Characteriscs
This is the product informaon document which is made
available to all prescribing physicians in the EU for
marketed products.
SUSAR
Suspected Unexpected
Serious Adverse Reacons
Suspected Unexpected Serious Adverse Reacon is the
term used to refer to an adverse event that occurs in a
clinical trial subject, which is assessed by the sponsor and
or study invesgator as being unexpected, serious and as
having a reasonable possibility of a causal relaonship with
the study drug.
TESS
CTIS Training Environment
Support Service
VHP
Voluntary Harmonisaon
Procedure
XEVPRM
eXtended EudraVigilance
Medicinal Product
Report Message
